Evoluţia pe termen lung a unui caz de angioedem prin deficit dobândit de C1-inhibitor esterază – particularităţi clinice şi terapeutice
Long‑term follow‑up of a case of recurrent angioedema due to acquired C1‑inhibitor deficiency – clinical and therapeutic aspects
Abstract
Angioedema (AE) is a frequent pathology in medical practice, raising significant diagnostic and therapeutic problems. Angioedema due to acquired C1-inhibitor esterase (C1-INH) deficiency is a rare form of AE without urticaria, with life-threatening potential outcome, often associated with lymphoproliferative or autoimmune diseases.We report the long-term outcome of an 80-year-old female patient, diagnosed in 2014 with AE due to acquired C1-INH deficiency, with good response to attenuated androgens, who has been constantly monitored in our clinic for 12 years. The onset of AE attacks was six years before diagnosis confirmation, and was considered first induced by angiotensin-converting enzyme inhibitors (ACEIs) therapy, but she continued to have AE attacks after the discontinuation of ACEI. No underlying disease could be identified, despite extended medical evaluation. AE episodes stopped after the initiation of treatment with danazol in September 2014 and the C1-INH and C4 plasma levels became normal after one month. We have closely monitored the possible side effects of danazol and the occurrence of other clinical and laboratory manifestations. Danazol therapy was well tolerated and we could not find any causal disease for the C1-INH deficiency. During the last three years, moderate blood hypereosinophilia was recorded, with no clinical manifestations and clear cause, still under investigation.
Keywords
acquired C1inhibitor deficiencyblood hypereosinophiliadanazolrecurrent angioedemaRezumat
Angioedemul (AE) reprezintă o patologie relativ frecvent întâlnită în practica medicală, care poate implica probleme de diagnostic etiologic şi de abordare terapeutică. Angioedemul prin deficit dobândit de C1 inhibitor esterază (INH) este o formă rară de angioedem recurent fără urticarie, cu risc potenţial de fatalitate, care poate fi asociat bolilor limfoproliferative sau autoimune.Prezentăm evoluţia pe termen lung a unei paciente în vârstă de 80 de ani, diagnosticată în 2014 cu AE prin deficit dobândit de C1INH, cu răspuns favorabil la tratamentul profilactic cu androgeni atenuaţi, monitorizată constant de 12 ani în secţia noastră. Debutul episoadelor de AE a fost cu şase ani înaintea confirmării diagnosticului, cauza iniţială fiind considerată tratamentul cu inhibitorii enzimei de conversie a angiotensinei (IECA), dar AE a continuat după oprirea IECA. Nu s-a putut identifica o patologie hematologică sau autoimună asociată, în ciuda evaluării multidisciplinare. După iniţierea tratamentului cu danazol în septembrie 2014, atacurile de AE s-au remis, iar valorile C4 şi C1INH s-au normalizat după o lună. Nu am înregistrat efecte secundare notabile ale terapiei cu danazol şi nu am identificat o cauză posibilă a deficitului de C1INH. În ultimii trei ani, pacienta asociază hipereozinofilie moderată persistentă, fără manifestări clinice care pot indica afectare organică şi fără o cauză aparentă.
Cuvinte Cheie
angioedem recurentdeficit dobândit de C1‑inhibitor esterazădanazolhipereozinofilie sangvinăIntroducere
Angioedemul (AE) reprezintă o patologie relativ frecvent întâlnită în practica medicală, cu o prevalenţă raportată cuprinsă între 2% şi 20% şi care poate implica probleme de diagnostic etiologic şi de abordare terapeutică(1). Poate fi izolat sau asociat cu alte manifestări alergice, în special cu urticarie şi/sau cu manifestări extracutanate şi reprezintă o urgenţă medicală cu risc vital când localizarea este la nivelul laringelui, al limbii sau asociază anafilaxie.
Angioedemul se caracterizează prin tumefierea localizată, autolimitantă, a ţesutului subcutanat sau submucos (respirator sau gastrointestinal), având ca substrat creşterea temporară a permeabilităţii vasculare şi extravazarea plasmatică. Frecvenţa de apariţie a AE poate varia de la episoade unice la episoade recurente timp îndelungat, cu severitate variabila, influenţată de substratul etiopatogenic. Cea mai frecventă localizare este cea facială (periorbitar şi la nivelul buzelor), urmată de localizarea abdominală (prin edem mezenteric) şi/sau periferică (palmo-plantar, genital, alte segmente).
AE poate fi clasificat în dobândit şi ereditar sau, în funcţie de mecanismul de producere, în: alergic (mediat de histamină), nonalergic (mediat de bradikinină) şi idiopatic(2).
AE mediat de histamină este declanşat prin degranularea mastocitelor şi eliberarea masivă a mediatorilor mastocitari, prin reacţii de hipersensibilizare alergică – IgE mediate sau non-IgE mediate(3). AE mediat de bradikinină (BK) este cauzat de excesul localizat de bradikinină, produs prin activarea sistemului kalikreină-kinină sau prin blocarea degradării metabolice a BK. Blocarea metabolizării BK este responsabilă de apariţia AE indus de consumul de inhibitori ai enzimei de conversie a angiotensinei (IECA), iar excesul sau supraproducţia de BK este consecinţa deficitului de C1‑inhibitor esterază (INH), care poate fi ereditar sau dobândit. Abordarea clinică adecvată şi diagnosticul diferenţial între aceste forme de AE sunt esenţiale, deoarece tratamentul şi prognosticul lor diferă(3,4).
Caracteristicile esenţiale în orientarea diagnosticului către AE nonhistaminergic sunt: absenţa unor factori trigger alergici, localizarea variată (facială, periferică şi gastrointestinală), durata mai lungă a episoadelor de AE (24–72 h) şi absenţa urticariei. Când localizarea AE este limitată doar la nivelul tractul gastrointestinal, acesta poate mima un sindrom subocluziv manifestat prin dureri abdominale severe, greaţă, vărsături, uneori ascită, supunând pacientul unor intervenţii chirurgicale inutile(5).
Angioedemul prin deficit dobândit de C1INH (acquired angioedema – AAE)
AAE se asociază frecvent cu neoplazii hematologice (de exemplu, limfom, gamapatie monoclonală cu semnificaţie incertă – MGUS) şi cu prezenţa de autoanticorpi anti-C1INH. Se consideră că prezenţa patologiilor asociate este cauza principală a deficitului de C1INH, un alt aspect caracteristic AAE fiind deficitul de C1q(6).
Alte forme de angioedem dobândit la adult sunt:
-
Angioedemul indus de IECA, ca formă de manifestare a intoleranţei la această clasă terapeutică. Mecanismul intoleranţei la IECA nu este pe deplin cunoscut – se consideră că anumiţi factori de risc, genetici şi/sau de mediu, joacă un rol important. Episoadele de AE sunt frecvent localizate la nivel facial (buze şi limbă), dar pot fi localizate şi la nivelul căilor respiratorii superioare (laringe) şi pot să apară în orice moment al tratamentului cu IECA, ceea ce pune probleme de diagnostic diferenţial(7).
-
Angioedemul idiopatic nu este însoţit de urticarie şi nu pot fi identificaţi factori declanşatori alergici (alimente sau medicamente). Incidenţa raportată în populaţia generală este de 0,05% Dozarea complementului (C4 şi C1INH) este în limite normale, nu are predispoziţie genetică şi nu are localizare facială sau abdominală, iar mecanismele nu sunt cunoscute(8).
-
Forme rare de AE – sindromul Gleich a fost descris prima dată în 1984 de către Gleich, fiind o boala rară, inclusă în clasificarea largă a sindroamelor hipereozinofilice (HES). Asociază episoade repetate de AE nonalergic, eozinofilie sangvină, urticarie, febră, creştere în greutate şi prezenţa unui nivel seric crescut de IgM(9,10).
Prezentarea cazului
Prezentăm cazul unei paciente cu vârsta actuală de 80 de ani, evaluată prima dată în Compartimentul de alergologie al Spitalului Clinic Colentina în ianuarie 2014, pentru episoade repetate de angioedem periferic, debutate în 2008, la vârsta de 66 de ani. Diagnosticul iniţial stabilit ambulatoriu a fost de AE indus de tratamentul cu inhibitori ai enzimei de conversie a angiotensinei (IECA), dar episoadele de AE au continuat şi după întreruperea acestuia. Atacurile de AE erau dureroase, cu localizare variabilă, periferică (cervical, membre superioare, inferioare şi fesier) şi neînsoţite de urticarie sau simptome digestive. Frecvenţa atacurilor de angioedem s-a agravat de la trei-patru episoade pe an până la atacuri săptămânale, cu durată de cel puţin 72 de ore, în ultimul an anterior prezentării în secţia noastră. Exceptând HTA, pacienta nu are antecedente personale şi heredocolaterale semnificative, neagă alergii medicamentoase sau alimentare, iar în perioadele dintre episoadele de AE era asimptomatică. Investigaţiile efectuate iniţial, anterior internării, nu au putut stabili un diagnostic cert, fiind considerat AE idiopatic, dar dozarea C4 şi C1INH cantitativ efectuată după doi ani de la debut a arătat valori scăzute constant, indicând posibil angioedem ereditar.
Cu ocazia primei internări, am confirmat diagnosticul de AE prin deficit dobândit de C1-INH şi am reevaluat pacienta pentru o posibilă patologie care ar putea fi responsabilă pentru deficitul de C1-INH. Testele de sânge, evaluarea imagistică (CT – „full body”) şi examenul măduvei osoase au fost negative pentru boli autoimune, alergii, neoplazii solide sau hematologice, iar C4 şi C1INH s-au menţinut scăzute. Nu s-au putut efectua teste genetice pentru un posibil angioedem ereditar, pe care nu le‑am considerat necesare, având în vedere debutul la vârstă înaintată (66 de ani) şi absenţa istoricului familial de angioedem. Diagnosticul pozitiv a fost de AE recurent prin deficit dobândit de C1INH, de cauză necunoscută, şi am decis iniţierea tratamentului profilactic cu acid tranexamic, în acel moment nefiind disponibile alte substanţe farmacologice pentru profilaxia şi tratamentul acestor forme de AE (concentrat de C1INH, blocanţi ai receptorilor de bradikinină sau inhibitori selectivi de kalikreină).
Evoluţia a fost iniţial favorabilă, dar după trei luni de tratament episoadele de AE au reapărut, cel mai sever fiind în septembrie 2014, când a avut primul atac cu localizare facială şi laringiană (confirmat de examenul ORL). În timpul atacului sever am decis întreruperea acidului tranexamic şi iniţierea tratamentului cu androgeni atenuaţi (danazol), iniţial 400 mg/zi timp de o săptămână, cu scăderea treptată a dozelor până la 100 mg/zi. Episodul sever s-a remis în a treia zi de tratament, iar după o lună valorile C1INH şi C4 serice s-au normalizat. Ulterior, după şase luni, pacienta a decis din proprie iniţiativă întreruperea tratamentului pentru o perioadă scurtă, pentru a identifica doza minimă eficientă. După patru săptămâni de la întreruperea tratamentului cu danazol, dozarea de C1INH şi C4 seric a evidenţiat valori semnificativ scăzute, dar fără apariţia AE (figura 1). Pacienta a reluat tratamentul profilactic cu danazol la o doză minimă de 50 mg zilnic, cu evoluţie ulterioară favorabilă.
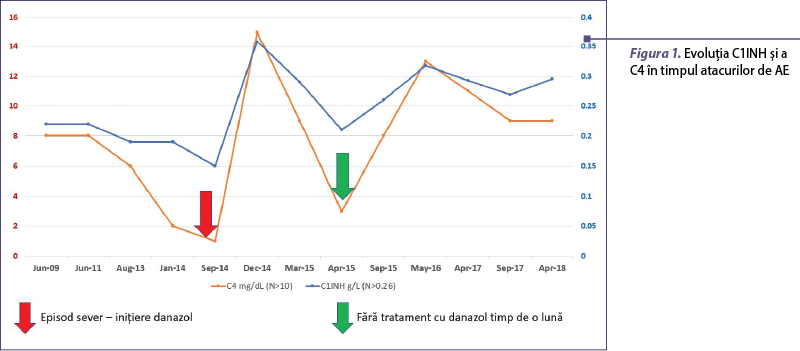
Pacienta a fost atent monitorizată şi evaluată periodic pentru a identifica posibile reacţii adverse ale tratamentului cu androgeni atenuaţi (în principal complicaţii cardiovasculare, metabolice şi tromboembolice), alte complicaţii sau boli concomitente.
Evoluţia AE
Eficacitatea tratamentului cu danazol a fost evaluată clinic şi prin dozarea periodică a complementului (C4) seric, care a avut valori normale la doze alternative (de la 50 mg administrat zilnic la 200 mg administrat de 2-3 ori/săptămână) – figura 2. Evoluţia clinică a fost favorabilă, fără atacuri de AE timp de opt ani de tratament cu androgeni atenuaţi, exceptând un episod recent (iunie 2021) de AE cu localizare periferică (picior şi gleznă stângă) după reducerea dozelor la 100 mg/săptămână, care s-a remis după recreşterea dozei de danazol la 200 mg zilnic pe perioada atacului. Ultima dozare de C1INH (antigen şi activitate), efectuată în noiembrie 2021, a fost în limite normale (0,312 g/L, respectiv 104%).
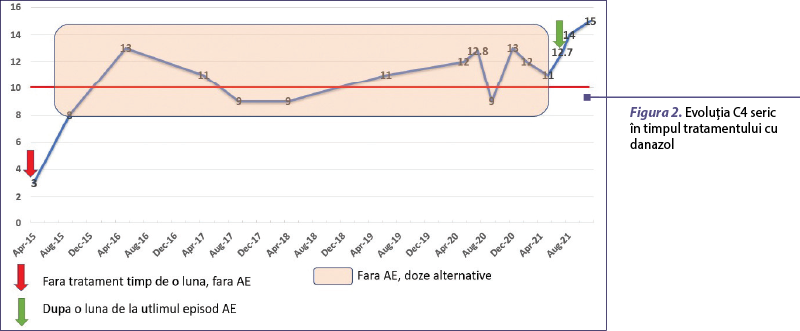
Evoluţia bolilor concomitente/asociate
După trei ani de tratament cu danazol pacienta a prezentat două episoade de abces pulmonar la nivelul lobului inferior stâng, prin infectarea unui chist pulmonar cunoscut (primul în 2017 şi recidivat după un an), remise cu antibioterapie de lungă durată, cu imagine chistică restantă de mici dimensiuni şi care nu au putut fi asociate tratamentului cu androgeni atenuaţi.
La evaluarea periodică, începând cu 2018, au fost decelate valori moderat crescute ale eozinofilelor sangvine (>1300/uL) – figura 3. Am investigat pacienta pentru o posibilă cauză secundară a eozinofiliei: infecţii parazitare, boli autoimune, boli hematologice, în special limfom şi gamapatie monoclonală, mastocitoză sistemică – toate analizele fiind normale. Proteina cationică eozinofilică (ECP) a avut valori moderat crescute la două determinări repetate, dar fără a prezenta simptome sugestive pentru afectare organică.
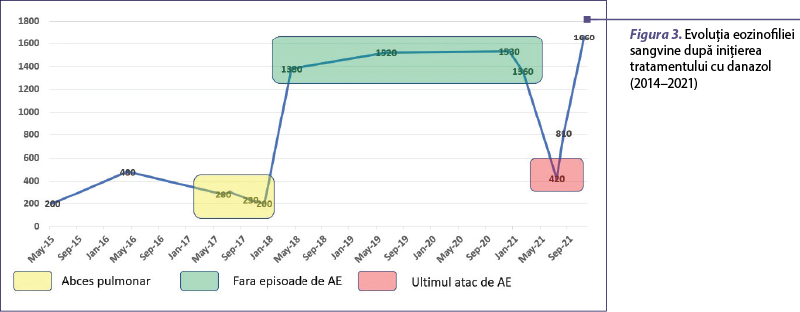
Discuţie
Angioedemul prin deficit dobândit de C1INH reprezintă un diagnostic dificil, stabilit în general după mulţi ani de atacuri repetate, cu evoluţie variabilă şi potenţial sever, frecvent asociat cu boli limfoproliferative. Informaţiile despre AE dobândit nonalergic la adult în practica medicală sunt de obicei limitate la formele induse de IECA. Dovezile conform cărora aproximativ 15% dintre pacienţii cu angioedem indus de IECA pot continua să aibă atacuri timp de câteva luni după întreruperea tratamentului cu IECA ar putea crea confuzie şi pot întârzia investigaţiile pentru alte cauze ale angioedemului dobândit(11).
Diagnosticul diferenţial al angioedemului dobândit (AAE) şi al celui ereditar (HAE)
Ambele forme de AE sunt considerate boli rare, având o prevalenţă estimată în populaţia generală de 1:50 000 în cazul HAE, respectiv 1:500 000 în cazul AAE. Spre deosebire de HAE, vârsta de debut în cazul AAE este mai înaintată, fiind afectaţi, de obicei, pacienţi cu vârsta de peste 40 de ani, fără antecedente familiale de AE şi care nu au prezente mutaţii ale genelor care codifică C1INH(12). De asemenea, fracţia complementului C1q este scăzută în AAE şi normală în HAE, ceea ce în cazul nostru este diferit. O analiză retrospectivă a unui lot de 77 de pacienţi diagnosticaţi cu AAE în perioada 1976–2015 a evidenţiat că dozarea C1q a avut valori normale la 19% dintre pacienţi(13).
Cea mai mare cohortă de pacienţi cu AAE a fost raportată în 2015, constând în 92 de cazuri, cu o urmărire medie de 4,2 ani de la diagnostic. Atacurile acute de AAE au fost localizate în principal la nivel facial şi abdominal şi jumătate dintre pacienţi au prezentat atacuri cu potenţial ameninţător de viaţă din cauza localizării la nivelul laringelui sau al limbii. Autorii au raportat o asociere remarcabilă a AAE cu limfomul indolent cu celule B (în special cu limfomul malign de zonă marginală) şi cu gamapatia monoclonală(14). O observaţie anterioară importantă menţionează că anomaliile imunologice subiacente şi angioedemul pot preceda cu câţiva ani expresia clinică a bolii maligne(15).
Tratamentul AAE ar trebui să ia în considerare boala de bază, precum şi frecvenţa şi severitatea angioedemului. Recomandările privind tratamentul recidivelor AAE este similar cu cel al HAE, folosind terapia de substituţie cu concentrat de C1-INH şi medicamente care intervin în metabolismul bradikininei (antagonişti ai receptorilor pentru bradikinină – icatibant, inhibitori de kalikreină – ecallantide), care în general nu sunt disponibile sau nu sunt aprobate pentru această indicaţie. Profilaxia pe termen lung a AAE se poate face cu androgeni atenuaţi sau agenţi antifibrinolitici precum acidul tranexamic, care este considerat de unii autori drept medicamentul de elecţie pentru AAE(16,17). Danazolul este un androgen sintetic, utilizat din 1976 în profilaxia pe termen lung a HAE şi reprezintă o opţiune utilă şi pentru AAE, datorită profilului bun de siguranţă şi cost-eficacitate. S-a dovedit că danazolul creşte nivelul seric al C1-INH atât în HAE, cât şi în AAE, deşi mecanismul precis nu este cunoscut în totalitate(18). Se consideră că danazolul reduce consumul de C1-INH în AAE şi creşte sinteza la nivel hepatic în HAE(19). Reacţiile adverse raportate au fost: creştere în greutate, semne de virilizare în cazul femeilor, dislipidemii, hipertensiune arterială, adenom hepatic şi risc uşor crescut de accident vascular cerebral şi infarct miocardic(20).
Asocierea angioedem – eozinofilie este sugestivă pentru diagnosticul de sindrom Gleich, însă în cazul de faţă eozinofilia a apărut după zece ani de la debutul simptomatologiei, atacurile de angioedem nu au fost însoţite de manifestări cutanate, iar deficitul de C1INH şi C4 seric decelat în timpul episoadelor de angioedem nu susţine acest diagnostic.
Particularităţile cazului prezentat sunt: întârzierea timp de şase ani a diagnosticului, localizarea predominant periferică a angioedemului, fără atacuri abdominale, având un singur episod cu localizare de gravitate (facial şi laringian), absenţa unei patologii subiacente timp de 12 ani de urmărire şi asocierea eozinofiliei sangvine.
Debutul AE la vârsta înaintată şi lipsa istoricului familial sau personal de angioedem nu susţin diagnosticul de HAE, iar absenţa reacţiilor de hipersensibilizare alergică şi a tratamentului cu IECA trebuie să indice evaluarea cauzelor care pot determina deficitul de C1-INH.
Concluzii
Cazul raportat este ilustrativ pentru diversele manifestări clinice şi pentru complexitatea angioedemului recurent în practica medicală. Diagnosticul, terapia pe termen lung şi monitorizarea pacienţilor cu angioedem recurent pot fi dificile şi trebuie să ia în considerare opinia experţilor şi recomandările actuale ale ghidurilor internaţionale.
Bibliografie
-
Bożek A, Zając M. Impact of comorbidities on risk of angioedema without urticaria in elderly patients. Allergy Asthma Clin Immunol. 2021 Dec 14;17(1):133. doi: 10.1186/s13223-021-00637-z.
-
Swanson TJ, Patel BC. Acquired Angioedema. 2021 Aug 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 28613639. Bernstein JA, Moellman J. Emerging concepts in the diagnosis and treatment of patients with undifferentiated angioedema. Int J Emerg Med. 2012;5(1):39
-
Crochet J, Lepelley M, Yahiaoui N, et al. Bradykinin mechanism is the main responsible for death by isolated asphyxiating angioedema in France. Clin Exp Allergy. 2019;49:252–254. doi:10.1111/cea.13297
-
Belbézier A, Bocquet A, Bouillet L. Idiopathic Angioedema: Current Challenges. J Asthma Allergy. 2020;13:137–144
-
Veronez CL, Campos RA, Constantino-Silva RN, Nicolicht P, Pesquero JB, Grumach AS. Hereditary Angioedema-Associated Acute Pancreatitis in C1-Inhibitor Deficient and Normal C1-Inhibitor Patients: Case Reports and Literature Review. Front Med (Lausanne). 2019 Apr 17;6:80. doi: 10.3389/fmed.2019.00080.
-
Bork K, Staubach-Renz P, Hardt J. Angioedema due to acquired C1-inhibitor deficiency: spectrum and treatment with C1-inhibitor concentrate. Orphanet J Rare Dis. 2019 Mar 13;14(1):65. doi: 10.1186/s13023-019-1043-3.
-
Leru PM, Anton VF, Bocşan C, Muntean A, Boda D. Acquired angioedema induced by angiotensin-converting enzyme inhibitors – experience of one hospital-based allergy center. Experimental and Therapeutic Medicine. 2020 Jul;20(1):68–72.
-
Leru PM, Marton C. Idiopathic angioedema – difficulties of clinical and therapeutic approach. Alergologia. 2020;4:176-178.
-
Mormile I, Petraroli A, Loffredo S, Rossi FW, Mormile M, Del Mastro A, Spadaro G, de Paulis A, Bova M. Episodic Angioedema with Hypereosinophilia (Gleich’s Syndrome): A Case Report and Extensive Review of the Literature. J Clin Med. 2021;10(7):1442.
-
Haber R, Chebl JA, El Gemayel M, Salloum A. Gleich syndrome: a systematic review. Int J Dermatol. 2020 Dec;59(12):1458-1465.
-
Faisant C, Armengol G, Bouillet L, Boccon-Gibod I, Villier C. Angioedema triggered by medication blocking the renin/angiotensin system: retrospective study using the French National Pharmacovigilance Database. J Clin Immunol. 2016;36:95–102.
-
Longhurst HJ, Zanichelli A, Caballero T, Bouillet L, Aberer W, Maurer M, Fain O, Fabien V, Andresen I, for the IOS Study Group. Comparing acquired angioedema with hereditary angioedema (types I/II): findings from the Icatibant Outcome Survey. Clin Exp Immunol. 2016;188:148–53.
-
Zanichelli A, Azin GM, Wu MA, Suffritti C, Maggioni L, Caccia S, Perego F, Vacchini R, Cicardi M. Diagnosis, course, and management of zngioedema in patients with acquired C1-Inhibitor deficiency. J Allergy clin Immunol Pract. 2017;5(5):1307-1313.
-
Gobert D, Paule R, Ponard D, Levy P, Fremeaux-Bacchi V, Bouillet L. A nationwide study of acquired C1-inhibitor deficiency in France: characteristics and treatment responses in 92 patients. Medicine. 2016;95:33.
-
Sheffer AL, Austen KF, Rosen FS, Fearon DT. Acquired deficiency of the inhibitor of the first component of complement: report of five additional cases with complementary on the syndrome. J Allergy Clin Immunol. 1985;75:640–6.
-
Agostoni A, Aygoren-Pursun E, Binkley KE, Blanch A, Bork K, Bouillet L. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114(3):51–131.
-
Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, Caballero T, Farkas H, Grumach A, Kaplan AP, Riedl MA, Triggiani M, Zanichelli A, Zuraw B, HAWK, under the patronage of EAACI (European Academy of Allergy and Clinical Immunology). Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014 May;69(5):602-16.
-
Leru PM, Anton VF, Bumbea H. Nine year follow-up of a rare case of angioedema due to acquired C1-inhibitor deficiency with late onset and good response to attenuated androgen. Allergy Asthma Clin Immunol. 2018;14:69. https://doi.org/10.1186/s13223-018-0274-5.
-
Kőhalmi KV, Veszeli N, Zotter Z. The effect of long-term danazol treatment on haematological parameters in hereditary angioedema. Orphanet J Rare Dis. 2016;11:18.
-
Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153–63.

Rezumate
Angioedemul (AE) reprezintă o patologie relativ frecventă în practica medicală, adresată medicilor alergologi, dar şi altor specialişti, în special serviciilor de urgenţă. Este considerat alergic în majoritatea cazurilor, dar poate ridica probleme de diagnostic şi de tratament, managementul fiind dificil în u...
Terapiile biologice în astmul sever – o perspectivă moleculară
Laura Haidar, Carmen Panaitescu
Astmul este o boală heterogenă definită printr‑un istoric de simptome respiratorii care variază în timp şi în intensitate, precum şi prin limitarea variabilă a fluxului de aer, care poate deveni persistentă. În ciuda unei game largi de tratamente disponibile pentru astm, 5-10% dintre pacienţi prezintă un răsp...
Alergenele moleculare ale fungului Alternaria alternata în diagnosticul alergologic de precizie
Florin-Dan Popescu, Mariana Vieru, Carmen Saviana Ganea, Lorena Mihaela Gheorghiţă, Irina Bucur, Nicolae Viorel Dumitrescu, Florica Popescu
Sensibilizarea mediată IgE la Alternaria alternata poate fi subestimată la pacienţii cu alergie respiratorie. Câteva alergene mole...
Mastocitoză cutanată asociată cu o formă rară de hipertensiune portală noncirotică – dificultăţi de evaluare diagnostică în practica clinică
Polliana Mihaela Leru, Vlad Florin Anton
Mastocitoza reprezintă un grup eterogen de boli rare, cu afectare cutanată şi multisistemică, fiind caracterizate prin activarea patologică sau acumularea de mastocite anormale în unul sau mai multe o...
Dificultăţi în evaluarea sindroamelor de activare mastocitară în practica clinică
Polliana Mihaela Leru, Vlad Florin Anton
Mastocitele (mast cells – MC) reprezintă celulele-cheie efectoare în reacţiile de hipersensibilitate sistemică şi locală şi sunt, de asemenea, implicate în diverse patologii inflamatorii acute sau cronice(1)....