Variability of response to multimodal therapy in a stage IV bronchial carcinoid: a case report
Variabilitatea răspunsului la terapia multimodală într-un carcinoid bronşic în stadiul IV: un raport de caz
Abstract
Pulmonary neuroendocrine neoplasms (NENs) are classified into neuroendocrine tumors (NETs) and neuroendocrine carcinomas (NECs). NETs comprise typical carcinoids (TC) and atypical carcinoids (AC), while NEC are classified into large-cell neuroendocrine carcinomas (LCNEC) and small-cell neuroendocrine carcinomas (SCLC). NETs and NECs are distinct lesions, without molecular overlap, and with different prognosis and therapeutic options. We present the case of a 65-year-old patient diagnosed with pulmonary AC, marking its debut with carcinoid syndrome in 2016. The peculiarity of this case is given by the secondary bone lesions in progression, highlighted by CT examinations, without expressing somatostatin receptors or increased metabolic activity, which leaves open the possibility of dedifferentiation or a metachronous tumor.Keywords
neuroendocrine tumoratypical carcinoidbone metastasesRezumat
Neoplasmele pulmonare neuroendocrine (NEN) se clasifică în tumori neuroendocrine (NET) şi carcinoame neuroendocrine (NEC). NET cuprind tumorile carcinoide tipice (TC) şi atipice (AC), în timp ce NEC sunt clasificate în carcinoame neuroendocrine cu celule mari (LCNEC) şi carcinoame neuroendocrine cu celule mici (SCLC). NET şi NEC sunt leziuni distincte, fără suprapunere moleculară şi cu prognostic şi opţiuni terapeutice diferite. Prezentăm cazul unui pacient de 65 de ani, diagnosticat cu AC pulmonar, debutul fiind prin sindrom carcinoid în anul 2016. Particularitatea acestui caz este dată de leziunile secundare osoase în progresie, evidenţiate la examinările CT, fără a exprima receptori de somatostatină sau activitate metabolică crescută, ceea ce lasă deschisă posibilitatea dediferenţierii sau a unei tumori metacrone.Cuvinte Cheie
tumoră neuroendocrinătumoră carcinoidă atipicămetastaze osoaseBackground
Pulmonary neuroendocrine neoplasms (NENs) are classified into neuroendocrine tumors (NETs) and neuroendocrine carcinomas (NECs). NETs encompass typical carcinoids (TC) and atypical carcinoids (AC), while NECs comprise large cell neuroendocrine carcinoma (LCNEC) and small cell carcinoma (SCLC). NETs and NECs are distinct lesions, without molecular overlap and with different prognosis and therapeutic options. The criteria for diagnosis include mitotic count per mm2 and necrosis, alongside a wide array of cellular and architectural features fulfilling neuroendocrine morphology(1). The most important distinction is between well differentiated tumors (NETs) of low to intermediate grade (TC and AC, corresponding to G1 and G2, respectively), and poorly differentiated neuroendocrine carcinomas (NECs, corresponding to G3), either SCLC or LCNEC(2). It is important to note that the defining criteria are effective enough to highlight low and intermediate grade, while failing to a large extent in the behavioral distinction of NECs, therefore the morphological approach to classification complies with a three-tier spectrum of clinical outcomes, survival rates and therapy options(1,3). Basically, pulmonary neuroendocrine tumors are currently considered to consist of three grades of malignancy, ranging from TC and AC to LCNEC and SCLC(4).
Overall, NE neoplasms account for approximately 20% of lung primary tumors, with SCLC accounting for approximately 15%, carcinoids for approximately 2% (with typical to atypical ratio of 10:1), and LCNEC for approximately 3%. Notably, studies in certain countries have shown a decrease in the incidence of SCLC over the last few decades due to a decrease in cigarette smoking, whereas the incidence of carcinoids has substantially increased, likely due to increased use of computed tomography (CT) imaging(5). The incidence of LCNEC has also increased, which may reflect a better recognition of this entity(6). Overall, lung carcinoids account for approximately 30% of NETs in the body. Notably, the vast majority of lung carcinoids are early-stage tumors, and only a minority of patients develop distant metastases(6,7).
Bronchial carcinoid tumors can either be asymptomatic or present wheezing, dyspnea, cough, hemoptysis and recurrent pneumonia due to bronchospasm and obstruction(8). The variability in clinical presentation may lead to delays in diagnosis or even to misdiagnosis. The differential diagnosis of a patient with symptoms of bronchial obstruction, bronchospasm and hemoptysis includes an obstructing bronchial carcinoma, endobronchial metastasis, hamartomas, asthma, aspirated foreign body, and chronic obstructive pulmonary disease(9).
TCs are mostly well-differentiated tumors of histologically ordered structure, larger than 5 cm, and exhibit <2 mitoses/10 high power fields (HPFs), usually without necrosis, while ACs have an atypical appearance with 2-10 mitoses/10 HPFs and a more aggressive tumoral profile, necrosis being present. They have a higher probability to metastasize, to recur, and a poorer overall prognosis(9,10).
The patients with AC are usually in a more advanced stage at diagnosis than the patients with TC. AC has a higher probability for developing micrometastases and therefore has a more aggressive clinical behavior. Even small tumors located in the periphery of the tracheobronchial tree can metastasize, suggesting that nodal micrometastases do not correlate with tumor size and stage. Immunohistochemical detection of micrometastases using chromogranin A and CK as markers allows for a more accurate staging(11).
Atypical carcinoids have been reported having an association with tobacco use, unlike typical carcinoids, where there is no known association to tobacco use or exposure to other carcinogens(12,13). Carcinoid syndrome, which is characterized by excessive serotonin release from tumors, leading to hot, red flushing of the face, severe and debilitating diarrhea and asthma attacks, is rarely associated with bronchial carcinoids(14).
Case presentation
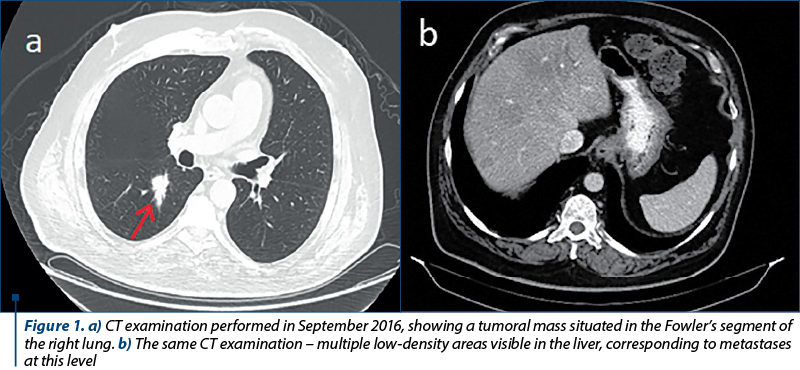
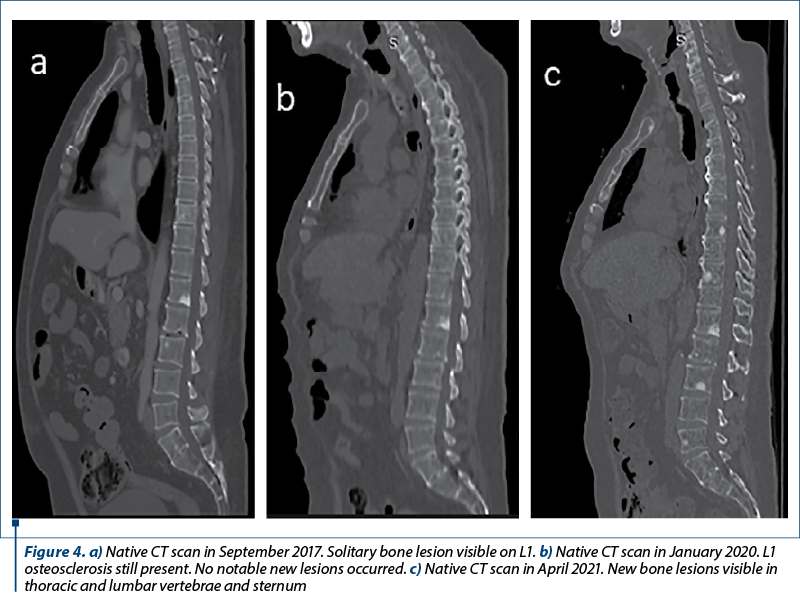
We present the case of a 62-year-old Romanian male patient with a medical history of high blood pressure and type 2 diabetes mellitus. His first symptoms began in August 2016, consisting of carcinoid syndrome (skin rash and diarrhea). An initial ultrasound examination revealed multiple hypoechoic formations in the liver. In order to obtain a superior view over these lesions, an MRI examination was performed, which confirmed the hepatomegaly with the presence of multiple lesions located throughout the liver, especially in the right lobe. They varied in size from a few millimeters to 27/23 mm at most. Also, a CT performed one month later found a tumoral mass in the Fowler’s segment of the right lung (Figure 1a) along with multiple lymphadenopathies situated in the pulmonary hilum, mediastinum and axilla, as well as an osteosclerotic lesion located in the vertebral body L1. The hepatic lesions were also present on the CT examination (Figure 1b). No cerebral lesions were reported.
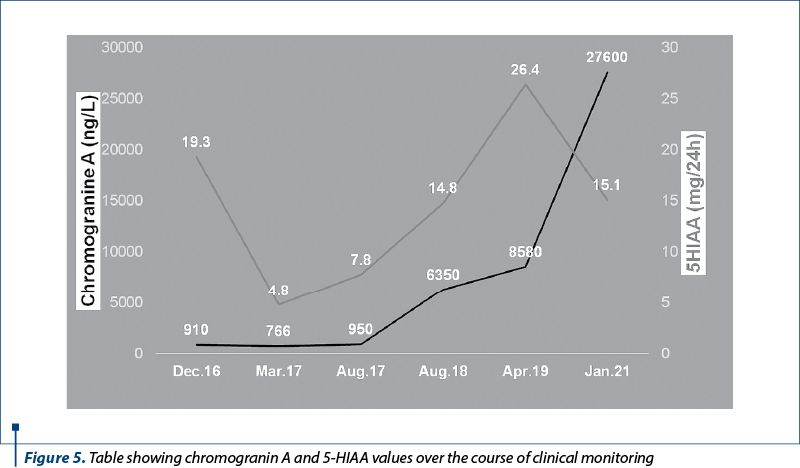
A bronchoscopy performed in October 2016 turned out negative, the colonoscopy and endoscopy results were normal, and the tumoral markers carcinoembryonic antigen (CEA), CA 19-9 and alpha-fetoprotein (AFP) were also within normal values range. Because the bronchoscopy was inconclusive, a liver biopsy was performed in order to clarify the diagnosis. It determined the presence of a neuroendocrine differentiation, but no immunohistochemistry (IHC) could be performed because of necrotic areas present within the biopsy sample. At that time, chromogranin A value was elevated (910 ng/l), serotonin was slightly above limit (265 µg/l) and 5-hydroxyindoleacetic acid (5-HIAA) value was also increased (19.3 mg/24 h).
Considering the neuroendocrine differentiation discovered in the liver and the elevated tumoral markers, the treatment began in November 2016, with octreotide (30 mg every 28 days). The follow-up CT examination in March 2017 showed stationary disease, but the next CT scan from September 2017 revealed progression, and for that reason the treatment was changed to capecitabine and temozolomide (second-line chemotherapy). In November 2017, the medical commission agreed upon the lung tumoral mass being the primary tumor.
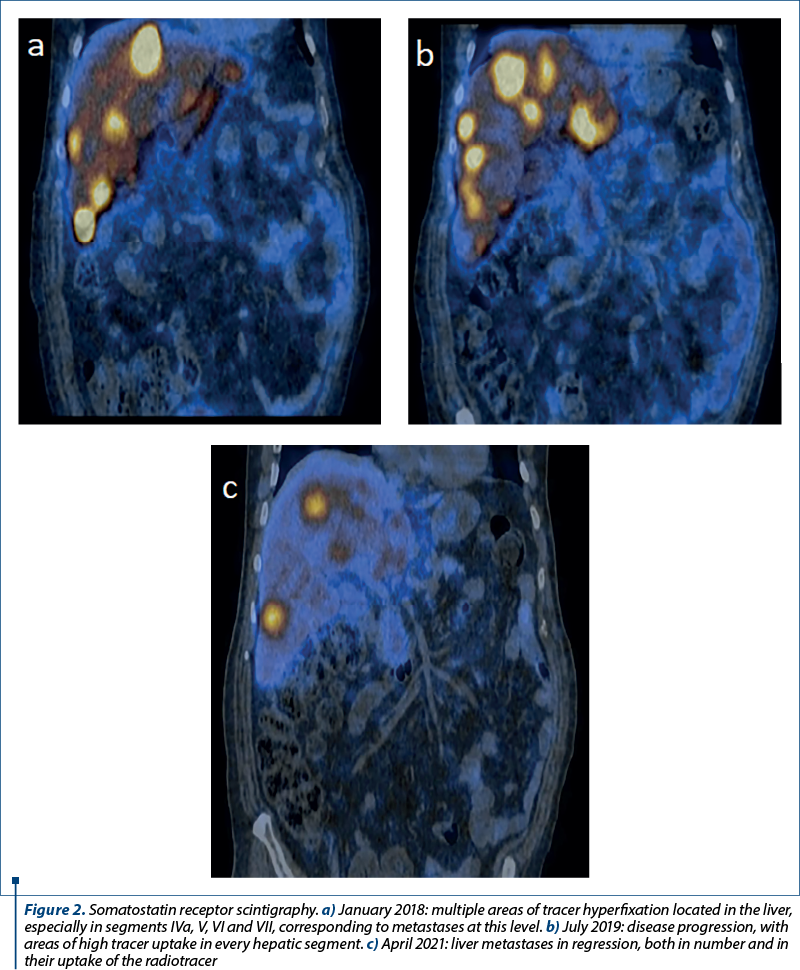
The first somatostatin receptor scintigraphy was performed in January 2018, both the primary tumor and the liver metastases expressing a high density of somatostatin receptors (Figure 2a). Therefore, octreotide was added again as a treatment, besides the second-line chemotherapy drugs. Chromogranin A levels rose continuously, reaching a value of 8580 ng/L in early 2019. The second somatostatin receptor scintigraphy was performed in July 2019 (Figure 2b), and due to further progression of the disease, as well as increased tumoral markers under treatment, the decision was made to change octreotide to lanreotide and recommend Lu-177-based peptide receptor radionuclide therapy (PRRT).
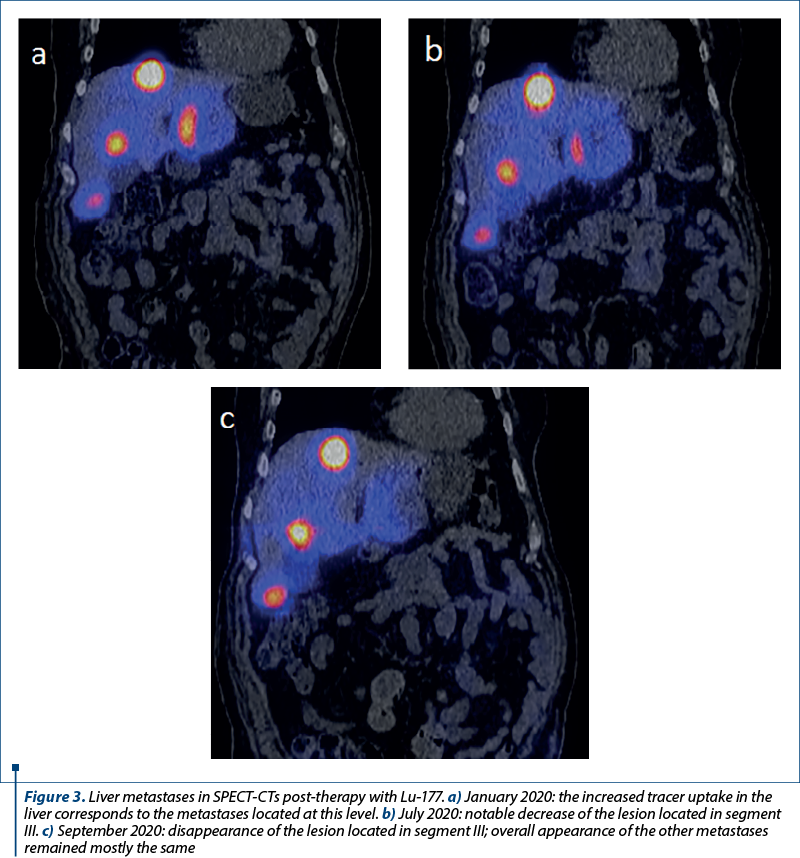
Four sessions of PRRT with Lu-177 were performed between November 2019 and September 2020. Post-therapy SPECT-CT images showed an intense uptake of Lu-177 in the hepatic lesions, largely unaffected by treatment, except for one located in the left lobe, which has reduced in size (Figure 3a-c). In December 2020, a CT scan evaluating the PRRT therapy showed mixed results: the primary tumor in the lung was in regression, the liver metastases were in slight progression, and the bone lesions were also in progression, rather unaffected by any treatment so far.
In order to initiate therapy with everolimus, the liver biopsy was repeated in March 2021, the result being neuroendocrine tumor G2. One month later, a third somatostatin receptor scintigraphy revealed a decrease in the primary tumor and hepatic metastases (Figure 2c). However, the bone lesions were still in progression (Figure 4a-c).
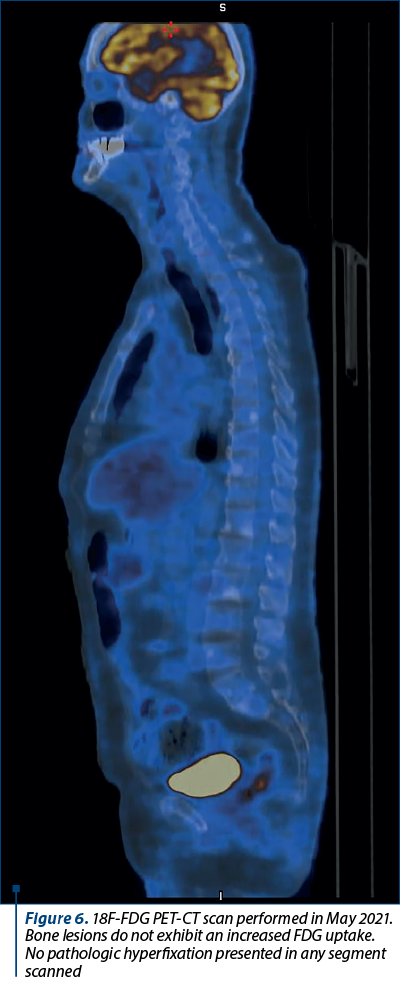
Chromogranin A value kept increasing, up to 27,600 ng/L in January 2021, while 5-HIAA had a peak in April 2019 (of 26.4 mg/24 h) and then decreased as a result of therapy (Figure 5). An 18F-FDG PET-CT scan was performed in May 2021, without any notable result (Figure 6). Also, PSA values were normal.
Discussion
Atypical carcinoid represents a rare lung neoplasm, included in the neuroendocrine spectrum and defined by Travis modified histological criteria(15,16). The intermediate prognosis in AC has been described in literature as being significantly improved in N0 patients compared to the N+ ones(17-19). Also, AC histology is an important prognostic factor related to survival of patients suffering from this disease(20-22). Due to the low incidence of pulmonary carcinoid tumors, there is a lack of large phase II or III trials regarding the treatment and management of pulmonary carcinoid tumors(14).
Surgery is the mainstay for the treatment of both typical and atypical bronchial carcinoids. In AC resection, lobectomy and pneumonectomy are the most common options, while with TC, bronchial sleeve resection which involves removal of less tissue solves the purpose since they are not as aggressive as AC(23). However, our patient could not undergo surgery because he already had liver and bone metastases at the time of diagnosis.
Somatostatin receptor scintigraphy and somatostatin receptor PET/CT prove to be more useful in imaging of well differentiated (G1) or moderately differentiated (G2) neuroendocrine tumors. A poorly differentiated tumor (G3) loses most of its original tissue characteristics (neuroendocrine), therefore it does not provide a good uptake of somatostatin analogues. However, as it evolves aggressively and the number of mitotic divisions increases, it becomes a candidate for evaluation in the 18F-FDG PET-CT examination(24).
A study conducted on 7334 patients in the Swedish Cancer Registry, all of them with neuroendocrine tumors, has shown that 25% of them had metastases. Of all patients with metastases, only 16% were bone metastases, the most common neuroendocrine tumors responsible being those of pancreas, small intestine and lung(25). Another study, from Italy, conducted on a group of patients with neuroendocrine lung neoplasms, determined that 31% had metastases, and of those, 42% presented bone lesions, bone being the second most common site of metastasis after the liver(26).
Two assumptions regarding the bone metastases were considered: the possible dedifferentiation from the primary neuroendocrine tumor, and the existence of a metachronous tumor responsible for the bone lesions. Prostate cancer was considered as a possible metachronous tumor due to its frequency in the general male population, as well as prostate cancer cells generally showing exquisite tropism for the bone(27-29). However, the patient’s PSA levels were normal. The 18F-FDG PET-CT examination could not confirm the existence of either the dedifferentiation or any metachronous tumor, prostatic or otherwise. Bone lesions did not present an increased uptake of the 18F-FDG radiotracer.
An investigation that would benefit the patient would be a PET-CT SSTR, which has a superior diagnostic performance compared to Octreoscan. One study has found a highly significant correlation between SSTR2A and SSTR5 and the SUVmax on the 68Ga-DOTA-NOC PET/CT scans to be concordant with the affinity profile of 68Ga-DOTA-NOC to the SSTR subtypes, therefore demonstrating the excellent qualification of somatostatin analogues in the diagnostics of NET(30). Also, several clinical studies have led to U.S. Food and Drug Administration approval of 68Ga-DOTATATE as an imaging agent for NET; the trials demonstrated high accuracy using histopathology or clinical follow-up for lesion verification and safety and a significant change regarding the management in a head-to-head comparison with Octreoscan(31-34). On the basis of its accuracy, SSTR PET/CT is already considered the standard of care in Europe and it is incorporated into clinical guidelines. SSTR radioligands further serve as predictive biomarkers to confirm target receptor expression and identify patients suitable for 177Lu-DOTATATE peptide receptor radionuclide therapy, currently under expanded access(31).
Conclusions
The peculiarity of this case is represented by the bone metastases that are most likely of neuroendocrine origin, but never expressed somatostatin receptors on scintigraphic examinations. Their evolution is different from both the primary tumor and the liver metastases, which had periods of response to therapy. The bone lesions did not progress favorably during any of the multiple treatment regimens, including the therapy with Lu-177 radiolabeled somatostatin analogue.
Conflicts of interests: The authors declare no conflict of interests.
Bibliografie
-
Metovic J, Barella M, Bianchi F, et al. Morphologic and molecular classification of lung neuroendocrine neoplasms. Virchows Arch. 2021;478(1):5-19. doi:10.1007/s00428-020-03015-z.
-
Rindi G, Klimstra DS, Abedi-Ardekani B, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. 2018;31(12):1770-1786. doi:10.1038/s41379-018-0110-y.
-
Travis W, Brambilla E, Burke A, Marx A, Nicholson A. WHO classification of tumours of the lung, pleura, thymus and heart. Lyon: IARC Press; 2015.
-
Skuladottir H, Hirsch FR, Hansen HH, Olsen JH. Pulmonary neuroendocrine tumors: incidence and prognosis of histological subtypes. A population-based study in Denmark. Lung Cancer. 2002;37(2):127-135. doi:10.1016/s0169-5002(02)00080-6.
-
Rekhtman N. Lung neuroendocrine neoplasms: recent progress and persistent challenges. Mod Pathol. 2022;35(Suppl 1):36-50. doi:10.1038/s41379-021-00943-2.
-
WHO Classification of Tumours Editorial Board. Thoracic Tumours. 5th ed. Lyon (France): International Agency for Research on Cancer. 2021.
-
Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134(11):1628-1638. doi:10.5858/2009-0583-RAR.1.
-
Quaedvlieg PF, Visser O, Lamers CB, Janssen-Heijen ML, Taal BG. Epidemiology and survival in patients with carcinoid disease in the Netherlands. An epidemiological study with 2391 patients. Ann Oncol. 2001;12:1295–300.
-
Garg R, Kumar R, Singh P, Kshetrimayum S. Atypical carcinoid tumor of the lung: A rare entity. Lung India. 2019;36(3):236-238.
-
Arrigoni MG, Woolner LB, Bernatz PE. Atypical carcinoid tumors of the lung. J Thorac Cardiovasc Surg. 1972;64:413–21.
-
Mineo TC, Guggino G, Mineo D, Vanni G, Ambrogi V. Relevance of lymph node micrometastases in radically resected endobronchial carcinoid tumors. Ann Thorac Surg. 2005;80:428–32.
-
Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119:1647–1651.
-
Corrin B. Neuroendocrine neoplasms of the lung. Curr Diagn Pathol. 1997;4:239–250.
-
Herde RF, Kokeny KE, Reddy CB, et al. Primary Pulmonary Carcinoid Tumor: A Long-term Single Institution Experience. Am J Clin Oncol. 2018;41(1):24-29. doi:10.1097/COC.0000000000000221.
-
Travis WD, Rush W, Flieder DB, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22(8):934-944.
-
Cañizares MA, Matilla JM, Cueto A, et al. Atypical carcinoid tumours of the lung: prognostic factors and patterns of recurrence. Thorax. 2014;69(7):648-653. doi:10.1136/thoraxjnl-2013-204102.
-
García-Yuste M, Matilla JM, Cueto A, et al. Typical and atypical carcinoid tumours: analysis of the experience of the Spanish Multicentric Study of Neuroendocrine Tumours of the lung. Eur J Cardiothorac Surg. 2007;31:192–7.
-
Cardillo G, Sera F, Di Martino M, et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77:1781–5.
-
Filosso PL, Rena O, Donati G, et al. Bronchial carcinoid tumors: surgical management and long-term outcome. J Thorac Cardiovasc Surg. 2002;123:303–9.
-
Thomas CF, Tazelaar HD, Jett JR. Typical and atypical pulmonary carcinoids: Outcome in patients presenting with regional lymph node involvement. Chest. 2001;119:1143–50.
-
Aydin E, Yazici U, Gulgosteren M, et al. Long-term outcomes and prognostic factors of patients with surgically treated pulmonary carcinoid: our institutional experience with 104 patients. Eur J Cardiothorac Surg. 2011;39:549–54.
-
Cao C, Yan TD, Kennedy C, et al. Bronchopulmonary carcinoid tumors: long-term outcomes after resection. Ann Thorac Surg. 2011;91:339–43.
-
Machuca TN, Cardoso PF, Camargo SM, et al. Surgical treatment of bronchial carcinoid tumors: A single-center experience. Lung Cancer. 2010;70:158–62.
-
Hofman MS, Hicks RJ. Changing paradigms with molecular imaging of neuroendocrine tumors. Discov Med. 2012;14(74):71-81.
-
Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. 2016;139(12):2679-2686.
-
Peri M, Botteri E, Pisa E, et al. A single-institution retrospective analysis of metachronous and synchronous metastatic bronchial neuroendocrine tumors. J Thorac Dis. 2018;10(7):3928-3939.
-
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492.
-
Ferlay JEM, Lam F, Colombet M, Mery L, Pineros M, Znaor A, Soerjomataram I, et al. Global cancer observatory: cancer today. Lyon, France: International Agency for Research on Cancer, 2020.
-
Wong SK, Mohamad NV, Giaze TR, Chin KY, Mohamed N, Ima-Nirwana S. Prostate Cancer and Bone Metastases: The Underlying Mechanisms. Int J Mol Sci. 2019;20(10):2587.
-
Kaemmerer D, Peter L, Lupp A, et al. Molecular imaging with ⁶⁸Ga-SSTR PET/CT and correlation to immunohistochemistry of somatostatin receptors in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2011;38(9):1659-1668.
-
Barrio M, Czernin J, Fanti S, et al. The Impact of Somatostatin Receptor-Directed PET/CT on the Management of Patients with Neuroendocrine Tumor: A Systematic Review and Meta-Analysis. J Nucl Med. 2017;58(5):756-761.
-
Haug AR, Cindea-Drimus R, Auernhammer CJ, et al. Neuroendocrine tumor recurrence: diagnosis with 68Ga-DOTATATE PET/CT. Radiology. 2014;270:517–525.
-
Haug AR, Cindea-Drimus R, Auernhammer CJ, et al. The role of 68Ga-DOTATATE PET/CT in suspected neuroendocrine tumors. J Nucl Med. 2012;53:1686–1692.
-
Deppen SA, Liu E, Blume JD, et al. Safety and efficacy of 68Ga-DOTATATE PET/CT for diagnosis, staging, and treatment management of neuroendocrine tumors. J Nucl Med. 2016;57:708–714.

A teaching experience in geriatric oncology: the Treviso SIOG advanced courses
Silvio Monfardini
Cancer predominantly affects older patients, but an integrated approach bringing together clinical oncologists and geriatricians for the best management of neoplasia in the elderly is not worldwide available. There is a need for some kind of shortcut that would teach the principles of geriatric oncology w...
Comparing oxidative metabolism in patients with uveal melanoma versus cutaneous melanoma
Mihai Adrian Păsărică, Paul Filip Curcă, Christiana Diana Maria Dragosloveanu, Valentin Dinu, Marian Burcea, Alexandru Grigorescu
Melanoamele maligne (MM) cutanate şi uveale prezintă caracteristici diferite de metastazare, fără un tratament sistemic standar...
Real-world and clinical trials for assessing the effectiveness of new drugs in oncology. The role of observational studies with exemplification in non-small cell lung cancer (NSCLC)
Mihaela Teodorescu, Alexandru Grigorescu
Datele din lumea reală (real-world data; RWD) şi studiile observaţionale (OS) sunt din ce în ce mai importante în cercetarea ...