Paucitatea ductelor biliare intrahepatice în practica pediatrică
Paucity of intrahepatic bile ducts in pediatric practice
Abstract
Neonatal cholestasis is the main cause of hospitalization in a pediatric hepatology department and represents the main indication for liver transplantation in children. Besides the biliary atresia, infectious, metabolic or toxic causes, one of the important causes of neonatal cholestasis is the paucity of intrahepatic biliary ducts. Alagille syndrome represents the association of this paucity of the biliary ducts with cardiac, vascular, renal, skeletal, ocular manifestations and characteristic facies. Alagille syndrome raises important issues of diagnosis, treatment and prognosis, which makes these aspects necessary to be known by pediatricians. The differential diagnosis from biliary atresia or other causes of biliary ducts paucity is important for the management of a patient with cholestasis. In the last period, besides the clinical diagnosis, there is an increasing importance of the genetic confirmation of Alagille syndrome in a patient with cholestasis, important for the prognosis of the evolution. Liver transplantation may be an effective solution for patients with Alagille syndrome, but the particularities of post-transplant evolution should be considered.Keywords
neonatal cholestasisbile duct paucityAlagille syndromeRezumat
Colestaza neonatală este principala cauză de internare într-un serviciu de hepatologie pediatrică şi reprezintă principala indicaţie de transplant hepatic la copil. Pe lângă cauzele malformative (atrezie de căi biliare), infecţioase, metabolice sau toxice, una din cauzele importante o reprezintă paucitatea ductelor biliare intrahepatice. Sindromul Alagille reprezintă asocierea acestei paucităţi a ductelor biliare cu manifestări cardiace, vasculare, renale, scheletale, oculare şi cu un facies caracteristic. Sindromul Alagille ridică probleme importante de diagnostic, tratament şi de prognostic, ceea ce face necesară cunoaşterea acestor aspecte de către medicul pediatru. Diferenţierea faţă de atrezia de căi biliare sau de alte cauze de paucitate de ducte biliare este importantă pentru managementul unui pacient cu colestază. În ultima perioadă, pe lângă diagnosticul clinic, o importanţă tot mai mare o are confirmarea genetică pentru încadrarea unui caz cu colestază în sindrom Alagille, important pentru prognosticul evoluţiei. Transplantul hepatic poate reprezenta şi în cazul unui pacient cu sindrom Alagille o soluţie eficientă, dar trebuie ţinut cont de particularităţile de evoluţie post-transplant.Cuvinte Cheie
colestază neonatalăpaucitate ducte biliaresindrom AlagilleColestaza intrahepatică poate avea drept cauze afecţiuni virale, metabolice, toxice sau poate fi secundară unor modificări ale integrităţii căilor biliare. Paucitatea (hipoplazia) ductelor biliare intrahepatice reprezintă o afecţiune caracterizată histologic de reducerea numărului ductelor biliare interlobulare. Poate fi prezentă la copii, dar şi la adulţi şi poate fi o afecţiune congenitală, moştenită sau dobândită. Paucitatea ductelor biliare poate fi definită doar histologic şi necesită biopsia hepatică dintr-un specimen care să conţină cel puţin cinci spaţii porte. Un raport între ductele bilare şi spaţiile porte sub 0,5 defineşte hipoplazia sau paucitatea de ducte biliare. Paucitatea de ducte biliare este cauză a 6,7% din cazurile de colestază intrahepatică(1-3).
În perioada neonatală, paucitatea de ducte hepatice poate avea două forme clinice: forma sindromatică (sindromul Alagille) şi forma non-sindromatică, descrisă în infecţii, boli metabolice, toxice, anomalii cromozomiale sau idiopatică(2,3).
I. Sindromul Alagille
Definiţie
Sindromul Alagille (cunoscut sub mai multe denumiri, cum ar fi paucitatea sindromatică a ductelor biliare intrahepatice, displazia arterio-hepatică sau hipoplazia biliară intrahepatică) este o boală genetică cu transmitere autozomal dominantă, dar cu expresie variabilă, care asociază colestază neonatală cu manifestări cardiace, scheletale, oculare şi facies caracteristic. Sindromul Alagille este o boală rară, cu o incidenţă de la 1:30.000 la 1:100.000 de nou-născuţi vii, reprezentând circa 5% din cazurile de colestază neonatală. Incidenţa iniţială, de 1:70.000, a sindromului Alagille părea a fi subestimată, având în vedere că incidenţa anterioară se referă doar la cazurile diagnosticate cu colestază neonatală şi nu sunt luate în considerare variabilitatea şi penetranţa redusă a acestei patologii(4-9). Acest sindrom a fost descris pentru prima dată în 1965 de către Smith, apoi în 1969 de Alagille(10,11).
Etiopatogenie şi genetică
În 1997 s-a demonstrat că sindromul Alagille este determinat de mutaţia unor gene care influenţează diferenţierea celulară şi dezvoltarea tisulară(12,13). Peste 90% dintre pacienţii cu sindrom Alagille prezintă mutaţia genei JAG1 (în unele studii până la 98%), situată pe braţul scurt al cromozomului 20 (20p12)(5,8,9,14-16). Pot exista cazuri (1-2% până la 2,5%) în care să existe mutaţii la nivelul NOTCH2, grup în care bolile renale par să fie mai frecvente(8,9,15,16). Gena JAG1 codifică proteina Jagged1, un ligant al proteinei Notch1 (proteină transmembranară cu rol asemănător factorului de creştere epidermal). JAG1 este exprimată la nivelul inimii şi rinichiului la adulţi, nu şi la nivel hepatic, dar este exprimată la nivelul ficatului fetal. În 50-70% din cazuri, mutaţiile sunt de novo; până la 50% din pacienţi au un părinte afectat(5,8,9,17).
Un screening genetic al pacienţilor cu sindrom Alagille ar cuprinde atât secvenţierea, cât şi analiza numărului copiilor, ceea ce se poate realiza prin secvenţiere Sanger şi MLPA sau prin NGS (Next Generation Sequencing)(16). În cazul unui pacient cu mutaţii JAG1 sau NOTCH2 dovedite, este recomandată testarea genetică şi a părinţilor(9).
Etiopatogenia sindromului Alagille nu este pe deplin elucidată. Se pare că există tulburări ale dezvoltării ductelor biliare intrahepatice, prin remodelarea plăcii ductale. Formarea ductelor biliare este inhibată, deoarece celulele precursoare cu potenţial dublu nu se dezvoltă în celule epiteliale biliare. Paucitatea de ducte biliare poate să nu fie prezentă în biopsia iniţială a multor sugari cu sindrom Alagille.
-
Manifestări clinice
Sindromul Alagille asociază în principal manifestări hepatice, cardiace, vasculare, renale, scheletale, oculare şi un facies caracteristic(5,18). În tabelul 1 este prezentată frecvenţa cu care apar principalele manifestări clinice în sindromul Alagille, după studiile publicate de către mai mulţi autori(4,19-25).

-
Manifestări hepatice:
-
icter colestatic, scaune acolice şi urine hipercrome;
-
prurit sever (apare rar înainte de 3-5 luni, dar este prezent la majoritatea pacienţilor în al treilea an de viaţă, chiar şi la pacienţi anicterici)(18);
-
hepatomegalie, splenomegalie;
-
ciroză hepatică;
-
insuficienţă hepatică;
-
hepatocarcinom.
-
-
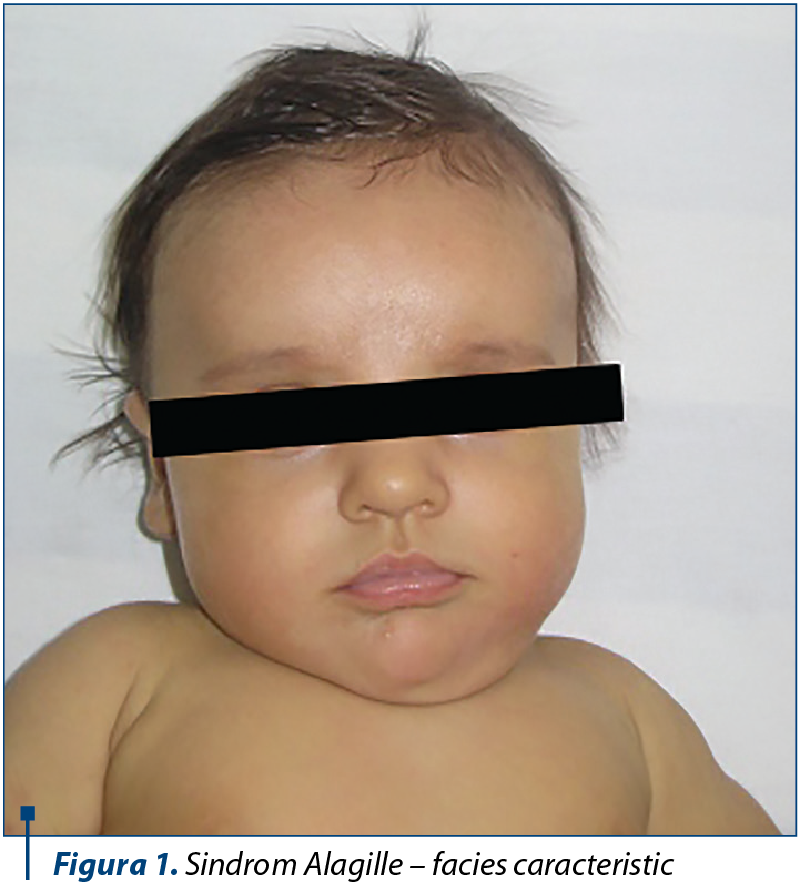
Facies caracteristic (poate să nu fie evident din primele luni de viaţă, fiind mai uşor de identificat cu creşterea vârstei): frunte largă, ochi înfundaţi, hipertelorism uşor, nas drept, bărbie ascuţită.
-
Modificări scheletale:
-
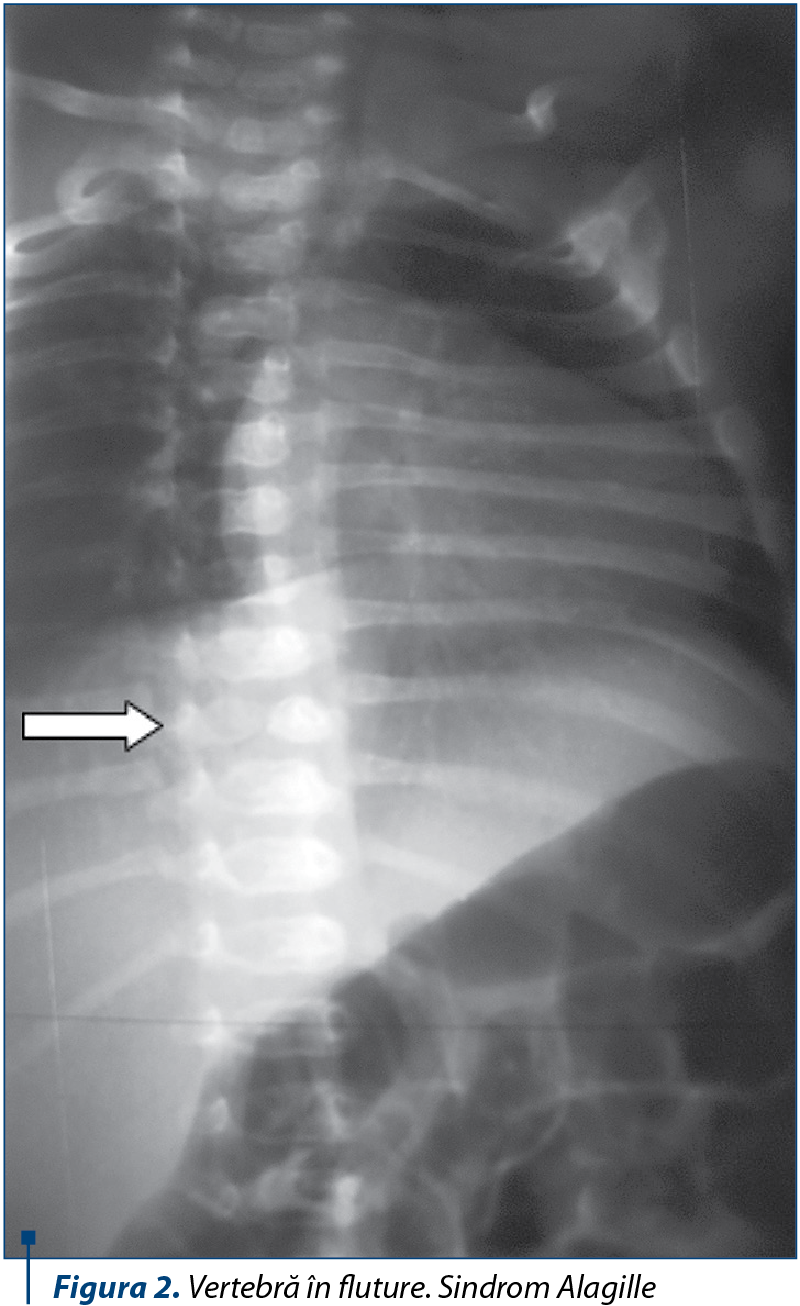
vertebră în „fluture” la nivel toracic (lipsa fuziunii la nivelul arcului anterior al corpului vertebral), dar fără repercusiuni asupra staticii coloanei vertebrale. Acest aspect poate apărea şi în alte sindroame: velo-cardio-facial, VATER, Crouzon, Jarcho-Levin şi disostoza spondilo-costală(5);
-
spina bifida ocultă;
-
falange distale scurte;
-
clinodactilie deget V;
-
ulna scurtă.
-
-
Modificări oculare:
-
embriotoxon posterior (proeminenţă anormală a liniei Schwalbe = unirea membranei Descemet cu uveea la unghiul camerei anterioare); nu are influenţă asupra acuităţii vizuale şi poate apărea şi la persoane sănătoase în 5-15% din cazuri(26);
-
anomalie Axenfeld;
-
calcifieri în spaţiul extracelular al nervului optic;
-
pigmentare anormală a retinei;
-
strabism;
-
pupile ectopice;
-
hipotrofia discului optic cu sau fără modificări ale vaselor retiniene.
-
-
Manifestări cardiace şi vasculare:
-
stenoza periferică a arterei pulmonare (izolată sau combinată cu alte anomalii cardiace);
-
hipoplazia severă a ramurilor arterei pulmonare;
-
tetralogie Fallot;
-
stenoza valvelor pulmonare;
-
stenoza aortică;
-
defect de sept ventricular;
-
defect de sept atrial;
-
anomalii de întoarcere venoasă.
-
-
Manifestări renale:
-
tulburări ale funcţiei de concentrare a urinei;
-
litiază renală;
-
anomalii structurale (rinichi mici, rinichi unic congenital, boală chistică renală);
-
nefropatie membranoasă;
-
mezangiolipidoză;
-
acidoză tubulară renală.
-
-
Deficit de creştere asociat şi retardului de creştere intrauterină.
-
Malnutriţie severă în cadrul sindromului Alagille sau secundar malabsorbţiei lipidelor şi refluxului gastroesofagian.
-
Alte manifestări vasculare: vasculopatie intracraniană (boala Moyamoya), anomalii vasculare – coarctaţie de aortă.
-
Alte manifestări clinice, mai rare: pubertate întârziată, hipogonadism, voce modificată, diateză hemoragică, neuropatie periferică, hipotiroidism, retard mintal, dificultăţi de învăţare, comportament antisocial.
Diagnostic
Diagnosticul sindromului Alagille se bazează mai ales pe caracteristicile clinice ale sindromului, la care se adaugă investigaţii paraclinice, inclusiv biopsia hepatică. Iniţial pentru diagnostic era necesară prezenţa paucităţii de ducte biliare, asociată cu cel puţin trei din cele cinci criterii majore: colestază, facies caracteristic, anomalii vertebrale, anomalii oculare şi suflu cardiac(7). În cazul debutului cu colestază în perioada neonatală, este necesar diagnosticul diferenţial cu atrezia de căi biliare.
Biochimic se caracterizează prin hiperbilirubinemie conjugată importantă (30xVN), care se poate ameliora odată cu creşterea în vârstă, şi prin creşterea acizilor biliari serici (100xVN). Transaminazele şi fosfataza alcalină pot fi foarte crescute (chiar peste 10xVN), dar ulterior pot să scadă, iar gGT este crescută iniţial (chiar şi la 20xVN). Colesterolul şi trigliceridele pot fi foarte crescute în formele severe de colestază. Funcţia de sinteză hepatică este păstrată, dar se poate instala insuficienţa hepatică în evoluţia severă (10-50% din cei cu manifestări hepatice în primul an de viaţă)(5,18).
Ecografia abdominală poate releva un colecist de dimensiuni reduse, contractat.
Scintigrafia hepatobiliară poate arăta eliminarea trasorului la nivel duodenal, ceea ce diferenţiază sindromul Alagille de atrezia de căi biliare, dar excreţia poate fi întârziată sau poate lipsi în cazul colestazei intrahepatice severe(18).
Biopsia hepatică poate evidenţia până la vârsta de 3 luni colestază, balonizare şi transformare acinară a hepatocitelor, cu formare de celule gigante (hepatită cu celule gigante), cu ducte biliare normale ca număr, dar cu elemente de distrucţie. Mai târziu, colestaza se menţine, spaţiile porte prezintă fibroză şi numărul ductelor biliare scade(27). Este necesară prelevarea de ţesut hepatic suficient încât să poată fi evaluate cel puţin 10 spaţii porte. În mod normal trebuie să existe un raport ducte biliare/spaţii porte de cel puţin 0,8. Paucitatea ductelor biliare se defineşte prin valoarea raportului ducte biliare/spaţiu port sub 0,5(6,28). La prematuri nu se aplică aceste reguli, deoarece numărul ductelor biliare este mai scăzut decât la nou-născutul la termen(27). În perioada neonatală, în mod normal nu se evidenţiază fibroză periportală şi centrolobulară, dar în 15-20% din cazuri poate exista evoluţie spre ciroză biliară. În unele cazuri, diagnosticul diferenţial histologic cu atrezia biliară este dificil de realizat. Paucitatea ductelor biliare poate fi dovedită histologic la doar 60% din pacienţii cu vârsta sub 6 ani şi la 95% din cei care au avut biopsia efectuată după 6 luni(18,21).
Diagnosticul genetic pune în evidenţă prezenţa mutaţiilor caracteristice la peste 90% din pacienţii cu manifestări clinice caracteristice. Prin tehnica FISH (hibridizare in situ cu fluorescenţă) se pot identifica deleţii ale genei JAG1 la 5-7% din pacienţi(29). Există puţine dovezi în ceea ce priveşte existenţa unei corelaţii genotip-fenotip în ceea ce priveşte mutaţiile JAG1 şi sindromul Alagille(8).
Tratament
Tratamentul pacienţilor diagnosticaţi cu sindrom Alagille depinde de manifestările clinice prezente şi de severitatea acestora. În colestaza severă se aplică măsuri suportive, cu o atenţie specială pentru tratamentul pruritului, şi suport nutriţional, cu suplimentarea vitaminelor A, D, E şi K. Tratamentul pruritului în sindromul Alagille se poate face cu colestiramină, rifampicină şi acid ursodeoxicolic, cu răspuns favorabil la circa 50-80% din pacienţi. S-a încercat şi utilizarea de fenobarbital, dar cu rezultate dezamăgitoare(6). În cazul hipercolesterolemiei se indică o dietă modificată corespunzător şi tratament cu colestiramină. În cazul anomaliilor cardiace şi renale asociate, se recomandă tratament specific. Este recomandată evitarea traumatismelor, mai ales craniene, având în vedere posibilele anomalii vasculare cu risc de hemoragie intracraniană(30,31). Mutaţiile la nivelul genei JAG1 pot avea rol în modificarea integrităţii endoteliului vascular(29). Examenul de fund de ochi este util pentru monitorizarea de rutină, fiind posibile episoade de hipertensiune intracraniană. S-a încercat uneori cu succes intervenţia chirurgicală pentru diversia biliară parţială externă, înainte de efectuarea transplantului hepatic(8,32).
Transplantul hepatic este indicat în sindromul Alagille la circa 20-50% din cei cu afectare hepatică în perioada de sugar(20-22,25,33). Următoarele situaţii pot reprezenta indicaţie de transplant hepatic: insuficienţa hepatică, ciroza hepatică, hipertensiunea portală, pruritul foarte intens fără răspuns la medicaţie, xantomatoza, deficitul sever de creştere, fracturi osoase(31). În general, modificările de fibroză sau ciroză, leziunile focale şi hipertensiunea portală importantă sunt mai rar întâlnite în sindromul Alagille comparativ cu atrezia de căi biliare, la momentul transplantului hepatic(33). Transplantul hepatic la copiii cu sindrom Alagille prezintă un risc crescut de complicaţii extrahepatice. Având în vedere frecvenţa complicaţiilor vasculare şi renale post-transplant, sunt necesare evaluarea atentă a sistemului vascular înainte de efectuarea transplantului şi monitorizarea tensiunii arteriale şi a imunosupresiei post-transplant(25,30).
Prognostic
Prognosticul depinde de severitatea manifestărilor hepatice (mai sever la cei cu colestază neonatală), cardiace şi renale(34). Majoritatea pacienţilor cu sindrom Alagille au evoluţie favorabilă. Nivelul ridicat al bilirubinei totale şi conjugate şi cel al colesterolului la copiii sub 5 ani sunt consideraţi factori de prognostic pentru asocierea afectării severe hepatice ulterioare. Afectarea cardiacă se asociază cu mortalitate în perioada de sugar, iar complicaţiile hepatice se asociază cu mortalitatea mai tardivă(32).
În 20-30% din cazuri, evoluţia este spre deces prin complicaţii determinate de boala cardiacă severă, ciroza hepatică cu insuficienţă hepatică şi infecţii.
Peste 20% dintre pacienţi au nevoie de transplant hepatic, din cauza colestazei severe şi a evoluţiei spre ciroză hepatică. Supravieţuirea post-transplant comparativ cu copiii cu atrezie de căi biliare este mai redusă la cei cu sindrom Alagille: 87% versus 96% la 1 an şi 86% versus 94% la 5 ani(25). Copiii cu sindrom Alagille care supravieţuiesc după transplant recuperează deficitul de creştere mult mai bine decât cei cu atrezie de căi biliare, demonstrând că acest deficit de creştere întâlnit în sindromul Alagille poate constitui indicaţie de transplant hepatic(25).
Este posibilă evoluţia spre hepatocarcinom (chiar şi în lipsa cirozei şi la vârste mici de 2-4 ani), hepatoblastom, hiperplazie focală nodulară şi steatoză(32,34).
II. Paucitatea de ducte biliare nonsindromatică
Paucitatea ductelor biliare poate fi prezentă într-un număr crescut de boli, fără a fi însoţită de modificările caracteristice sindromului Alagille. În aceste cazuri, paucitatea ductelor biliare poate fi evidentă histologic chiar din prima săptămână de viaţă.
Cauze de paucitate nonsindromatică a ductelor biliare la sugari:
-
Prematuritate.
-
Hepatita neonatală idiopatică severă.
-
Infecţia congenitală (citomegalovirus, rubeolă, sifilis, hepatită B).
-
Boli metabolice (deficit de a1-antitripsină, fibroză chistică, sindrom Zellweger, colestaza intrahepatică familială progresivă 1, sindrom Ivemark, sindrom Prune-belly, hipopituitarism, mutaţie HNF-1ß).
-
Anomalii cromozomiale (trisomie 18, 21, trisomie parţială 11, monosomie X).
-
Boli imune – boala grefă-contra-gazdă (transplant de măduvă, transplant de celule stem în perioada neonatală).
-
Paucitate de ducte biliare idiopatică (posibil familială).
În lotul descris de Yehezkely-Schildkraut în Israel, 7 din 10 sugari cu paucitate nonsindromatică de ducte biliare au prezentat această caracteristică, asociată unei anumite patologii: câte doi pacienţi cu infecţie cu citomegalovirus şi colestază intrahepatică familială progresivă şi câte un pacient cu sindrom ARC, boala Niemann-Pick de tip C şi boală mitocondrială(35). Infecţia cu citomegalovirus pare a fi frecvent cauza paucităţii nonsindromatice de ducte biliare, fiind descrisă şi în 3 din cele 8 cazuri prezentate de DeTommasso(1).
Paucitatea de ducte biliare este asociată cu fibroza hepatică şi cu posibila evoluţie spre ciroză. Totuşi, în lotul descris de Yehezkely-Schildkraut, doar un singur pacient prezenta fibroză, spre deosebire de studiile lui Kahn et al., care au găsit fibroză în toate probele biopsiate(28,35).
Evoluţia nu poate fi prognozată în funcţie de vreun parametru clinic, biochimic sau histologic, dar asocierea bolilor metabolice cu paucitatea de ducte biliare poate avea un prognostic prost(35).
Având în vedere multitudinea circumstanţelor în care poate apărea, prognosticul cazurilor de paucitate nonsindromatică de ducte biliare variază şi pare a fi dependent de patologia la care se asociază. Se consideră că este necesară, din acest punct de vedere, clasificarea pacienţilor în două grupe: paucitate (hipoplazie) de ducte nonsindromatică secundară, la care etiologia s-a putut stabili, şi cei la care nu s-a putut defini etiologia, paucitate (hipoplazie) de ducte nonsindromatică primară.
Diferenţierea faţă de cauzele de colestază neonatală extrahepatică este foarte importantă pentru evitarea riscurilor chirurgicale. Această formă nonsindromatică nu trebuie considerată de la început ca având un prognostic prost, deoarece evoluţia favorabilă poate fi posibilă. Prognosticul infaust poate fi rezervat formelor primare sau idiopatice(1).
Conflict of interests: The author declares no conflict of interests.
Bibliografie
- DeTommaso AMA, Kawasaki AS, Hessel G. Paucity of intrahepatic bile ducts in infancy - experience of a tertiary center. Arq Gstroenterol. 2004; (41)3:190-2.
- Piccolli DA, Witzleben CL. Disorders of the biliary tract. Part I: intrahepatic bile ducts. In: Walker WA, Durie PR. Hamilton JR, et al. eds. Pediatric Gastrointestinal Disease. Philadelphia: BC Decker. 2000:895-914.
- Desmet JD. The cholangiopathies. In: Suchy FJ, Sokol RJ, Balistreri WF. eds. Liver Disease in Children. Philadelphia: Lippincott Williams & Wilkins. 2001:43-60.
- Alagille D. Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases.
- J Pediatr. 1987;110:195-200.
- Spinner NB. Genetics of Alagille syndrome. Progress in Pediatric Cardiology. 2005;20:169 – 176.
- MacMillan JC, Shepherd R, Heritage M. Arteriohepatic dysplasia (Alagille syndrome; Watson-Alagille syndrome). Baillieres Clinical Gastroenterology. 1998; 12 (2):275-291.
- Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34: 152-7.
- Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genetics. 2012;20:251-257.
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. The Application of Clinical Genetics. 2016;8:75-82.
- Smith DW, Optiz JM, Inhorm SL. A syndrome of multiple developmental defects including polycystic kidneys and intrahepatic biliary dysgenesis in two siblings. J Pediatr. 1965;67:617-24.
- Alagille D, Habib EC, Thomassin N. L’atresie des voies biliaires extrahepatiques permeables chez l’enfants. J Par Pediatr. 1969:301.
- Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997; 16 (3):243-51.
- Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat genet. 1997;16(3):235-42.
- Krantz ID, Piccoli DA, Spinner NB. Clinical and molecular genetics of Alagille syndrome. Curr Opin Ped. 1999;11:558-64.
- Kamath BM, Piccoli DA. Alagille syndrome. In: Liacouras CA, Piccoi DA (editors). Pediatric Gastroenterology. The Requisites in Pediatrics. Mosby Elsevier, Philadelphia, 2008.
- Gilbert MA, Bauer RC, Rajagopalan R, et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Human Mutation. 2019;1-24.
- Crosnier C, Driancourt C, Raynaud N, et al. Analysis of mutations of the Jagged1 gene in patients with Alagille syndrome: evidence for most cases being sporadic. Gastroenterology. 1999;116:141-8.
- Piccoli DA, Spinner NB. Alagille syndrome and the Jagged1 gene. Sem Liv Dis. 2001;21(4): 525-534.
- Deprettere A, Portmann B, Mowat AP. Syndromic paucity of the intrahepatic bile ducts: diagnostic difficulty; severe morbidity throughout early childhood.
- J Pediatr Gastroenterol Nutr. 1987;6:865-871.
- Hoffenberg EJ, Narkewitz MR, Sondheimer JM, et al. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr. 1995; 127:220-224.
- Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: requency and relation to prognosis. Hepatology. 1999;29:822-829.
- Quiros-Tejeira RE, Ament ME, Heyman MB, et al. Variable morbidity in Alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr. 1999;29:431-437.
- Garcia MA, Ramonet M, Ciocca M, et al. Alagille syndrome: cutaneous manifestations in 38 children. Pediatric Dermatology. 2005;22(1):11-14.
- Englert C, Grabhorn E, Burdelski M, et al. Liver transplantation in children with Alagille syndrome: indications and outcome. Pediatr Transplant. 2006;10:154-8.
- Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transplantation. 2012; 18: 940-948.
- Hingorani M, Nischal KK, Davies A, et al. Ocular abnormalities in Alagille syndrome. Ophtalmology. 1999;106:330-7.
- Waters BL, Blaszyk H. Diseases involving intrahepatic bile ducts. Curr Diag Pathology. 2005; 11:7-18.
- Kahn E, Daum F, Markowitz J, et al. Nonsyndromatic paucity of interlobular bile ducts: light and electron microscopic evaluation of sequential liver biopsies in early childhood. Hepatology. 1986;6:890-901.
- Mieli-Vergani G, Hadzic N. Biliary atresia and neonatal disorders of the bile ducts. In: Wyllie R, Hyams JS (editors). Pediatric Gastrointestinal and Liver Diseases. Pathophysiology, Diagnosis, Management. 3rd edition. Saunders Elsevier, Philadelphia 2006.
- Ganschow R, Grabhorn E, Helmke K, Rogiers X, Burdelski M. Liver Transplantation in Children with Alagille Syndrome. Transplantation Proceedings. 2001; 33,3608-3609.
- Maldini G, Torri E, Lucianetti A, et al. Orthotopic Liver Transplantation for Alagille Syndrome. Transplantation Proceedings. 2005; 37:1174–1176.
- Vajro P, Ferrante L, Paolella G. Alagille syndrome: an overview. Clinics and Research in Hepatology and Gastroenterology. 2012;36:275-77.
- Hwang SM, Jeon TY, Yoo SY, et al. Alagille syndrome candidates for liver transplantation: differentiation from end-stage biliary atresia using preoperative CT. PLoS ONE. 2016;11 (2):e0149681.
- Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001; 49:431-435.
- Yehezkely-Schildkraut V, Munichor M, Mandel H, et al. Nonsyndromic paucity of interlobular bile ducts: report of 10 patients. J Pediatr Gastroenterol Nutr. 2003; 37:546-549.

Particularităţi şi aspecte comune ale intoxicaţiilor în populaţia pediatrică şi cea adultă
Roxana Mihaela Barbu, Maria Cristina Gavrilescu, Bogdan A. Stana, Victoria Paraschiva Luca
Intoxicaţiile acute la grupa de vârstă pediatrică reprezintă una dintre principalele cauze de mortalitate şi de morbiditate, cu impact major în ceea ce priveşte mortalitatea prematură şi alterarea cal...
Efecte locale ale AINS
Evelina Moraru, Alice Azoicăi
Antiinflamatoarele nesterodiene (AINS) reprezintă o verigă importantă în terapia antalgică şi antiinflamatorie, atât la adulţi, cât şi la copii. Mecanismele clasice de acţiune sunt completate recent de implicaţii ale sistemului endocanabinoid ca parte a efectului lor analgezic, iar studiile recente cu R-pro...
Boala Kawasaki – actualităţi în sprijinul practicianului pediatru
Bianca Simionescu
Boala Kawasaki este o vasculită sistemică acută, percepută încă uneori ca o boală exotică şi, în consecinţă, posibilitatea acestui diagnostic poate fi trecută cu vederea sau amânată. Deşi prima descriere a bolii Kawasaki a fost făcută în Japonia în urmă cu 50 de ani, diagnosticul pozitiv se bazează şi în prez...
Evaluarea funcţională a tractului digestiv inferior la copii – manometria anorectală
Daniela Pop, Radu Samuel Pop, Diana Cireap, Georgiana Dragomir, Dorin Farcău
Manometria anorectală este o metodă obiectivă utilizată pentru examinarea funcţiei anorectale şi a sensibilităţii rectale. Scopul acestui studiu a fost evaluarea principalelor indicaţii de efectuare a manometriei a...
Particularități evolutive la doi pacienți cu atrezie de căi biliare – prezentare de cazuri
Gabriel Benţa, Alina Grama, Tudor Lucian Pop
Atrezia biliară (ACB) face parte din categoria colangiopatiilor mediate imunologic, alături de colangita sclerozantă neonatală şi de colangita sclerozantă primitivă, afecţiuni inflamatorii progresive şi severe, care...