The increasing incidence of new cases of tuberculosis, of latent forms and with multiple and extended resistance, required the acceleration of the research and elaboration of the anti-tuberculosis drugs by elucidating new targets. The main directions of study of the new compounds focused on the influence on the cell wall, the synthesis of proteins and nucleic acids, the energy metabolism, as well as on the immune status of the host. At the base of these strategies were the data regarding the structural components and the particularities of the life cycle of mycobacteria at different stages of disease evolution.
CERCETARE
Direcţii de cercetare şi elaborare de preparate antituberculoase
Directions for research and development of anti-tuberculosis drugs
First published: 30 octombrie 2019
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/FARM.190.5.2019.2632
Abstract
Rezumat
Creşterea incidenţei cazurilor noi de tuberculoză, a formelor latente şi cu rezistenţă multiplă şi extinsă, a impus accelerarea cercetării şi elaborării preparatelor antituberculoase prin elucidarea unor ţinte noi. Direcţiile prioritare de studiu al compuşilor noi s-au axat pe influenţa asupra peretelui celular, sintezei proteinelor şi acizilor nucleici, metabolismului energetic, precum şi asupra statusului imun al gazdei. La baza acestor strategii au stat datele referitoare la componentele structurale şi particularităţile ciclului de viaţă al micobacteriilor la diferite etape de evoluţie a maladiei.
Tuberculoza (TBC) rămâne o problemă majoră de sănătate publică la nivel mondial, fiind principala cauză a decesului determinat de un singur agent infecţios (cu excepţia infecţiei HIV/SIDA). Datele epidemiologice demonstrează că TBC are o incidenţă destul de mare, în ciuda eforturilor depuse în vederea profilaxiei şi tratamentului. Astfel, Organizaţia Mondială a Sănătăţii (OMS) estimează că numărul de cazuri noi şi de decese cauzate de TBC a constituit în 2013 – 9 milioane, respectiv 1,5 milioane; în 2014 – 9,6 milioane, respectiv 1,5 milioane; în 2015 – 10,4 milioane, respectiv 1,8-2 milioane; în 2016 – 10,4 milioane, respectiv 1,3 milioane; în 2017 – 10 milioane, respectiv 1,3 milioane, cu o creştere concomitentă a tulpinilor cu polirezistenţă şi rezistenţă extinsă de circa 480000-558000 de cazuri. Aproximativ 1,7 miliarde de oameni (23% din populaţia lumii) se estimează că au o infecţie latentă asimptomatică de TBC şi, prin urmare, sunt expuşi riscului de a dezvolta boala activă în timpul vieţii lor(8,13,16,21,23,24,28,29).
Datele de ultimă oră, pentru 2017, denotă că, din cele 10 milioane de persoane care au dezvoltat TBC în 2017 5,8 milioane au fost bărbaţi, 3,2 milioane femei şi 1 milion de copii. Cazuri de TBC au fost constatate în toate ţările şi la toate grupurile de vârstă, dar în general 90% erau adulţi (cu vârste ≥15 ani), 9% erau persoane infectate cu HIV (72% în Africa) şi două treimi erau din opt ţări: India (27%), China (9%), Indonezia (8%), Filipine (6%), Pakistan (5%), Nigeria (4%), Bangladesh (4%) şi Africa de Sud (3%). Acestea şi alte 22 de ţări din lista celor 30 de ţări cu pericol major pentru TBC au constituit 87% dintre cazurile mondiale. În regiunea europeană a OMS şi în regiunea OMS din America au fost estimate circa 3% dintre cazurile globale. Severitatea epidemiilor naţionale variază foarte mult în rândul ţărilor. În 2017, în majoritatea ţărilor cu venituri ridicate au fost circa 10 cazuri noi la 100 000 de locuitori, 150-400 în cele 30 de ţări cu risc major pentru TBC şi peste 500 în câteva ţări, inclusiv Mozambic, Filipine şi Africa de Sud(29).
Tuberculoza sensibilă la medicamente poate fi vindecată în decurs de 6-8 luni cu regimul de tratament standard, dar apariţia TBC cu rezistenţă multiplă şi extensivă la medicamente (MDR/XDR), al cărei tratament durează cel puţin 20 de luni, cu rezultate scăzute previzibile, reprezintă o mare ameninţare pentru sănătatea umană şi subliniază necesitatea de a descoperi şi de a dezvolta noi şi eficiente preparate antituberculoase. Cauza principală a tratamentului de durată este prezenţa micobacteriilor în stare inactivă, cu o activitate metabolică reversibilă. În timpul infecţiei, pacienţii pot găzdui trei subpopulaţii diferite de micobacterii: a) bacterii extracelulare în creştere activă care sunt prezente, de obicei, în interiorul cavităţilor aerate; b) bacili cu creştere intermitentă; c) bacili latenţi care sunt prezenţi în leziuni cu mediu acid şi în condiţii anaerobe, precum leziuni inflamatorii sau în macrofage, şi nu sunt afectaţi de terapia standard. Aceste subpopulaţii preexistente nu au o creştere şi prezintă o capacitate de a supravieţui expunerii la concentraţii mari de preparate. Ele pot rămâne inactive în timpul vieţii unui individ sau se pot resuscita în orice moment şi să progreseze spre tuberculoză activă, îndeosebi la pacienţii imunocompromişi: infectaţi cu virusul imunodeficienţei umane (HIV), cu diabet zaharat, vârstnici. Din acest motiv, obiectivul principal al cercetărilor de descoperire şi studiu al preparatelor antituberculoase constă în identificarea medicamentelor cu noi mecanisme de acţiune, cu potenţialul de a scurta durata terapiei şi a influenţa micobacteriile în creştere şi pe cele latente(16).
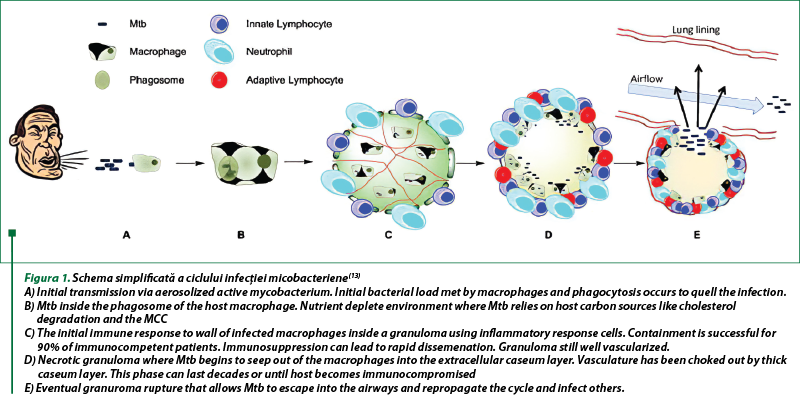
Infecţia iniţială apare atunci când un purtător transmite micobacteriile în aer prin tuse, cu un conţinut de picături mici cu un număr mic de microorganisme individuale (<10) şi care trebuie să fie inhalate adânc în plămâni. În acest moment, macrofagii-gazdă din plămân încearcă să fagocitoze patogenul (figura 1.3A) şi să-l transporte peste epiteliul alveolar şi în plămân. Aceasta declanşează un răspuns proinflamator care va recruta alte celule imune pentru a forma un granulom încapsulat, care este un răspuns imun tipic la un agent patogen. În acest moment, cea mai mare parte a încărcăturii bacteriene este conţinută în interiorul a ceea ce sunt acum considerate macrofage spumoase (figura 1B), care încep să contureze exteriorul granulomului. Când granulomul este iniţial format, acesta este bine vascularizat şi sunt prezente multe celule imune (figura 1C), care susţin capacitatea medicamentelor de a ajunge la infecţie şi sistemul imunitar al gazdei pentru a combate agentul patogen. Pe măsură ce granulomul continuă să se maturizeze împotriva răspunsului imun indisolubil, peretele exterior începe să se întărească cu o capsulă fibroasă groasă, iar miezul interior este îndoit de celulele imune. Spre deosebire de macrofagele spumoase, linia exterioară a acestei leziuni, celulele mor şi cazeumul se dezvoltă la nivelul miezului. În acest moment, granulomul este considerat necrotic, iar bacilii există extracelular în acest caz şi pot intra în starea lor în mare parte latenţi (figura 1D). Leziunile granulomului necrotic (cunoscute şi sub numele de tuberculi) fac ca penetrarea medicamentului să fie dificilă. După mulţi ani sau în caz de afectare a sistemului imunitar, granuloamele vor fuziona în căile respiratorii ale plămânilor şi vor elibera agentul patogen, cu răspândirea în ţesuturi noi şi gazde noi (figura 1E). Fiecare dintre aceste puncte din ciclul de viaţă al micobacteriilor are diferite micromedii care pot afecta susceptibilitatea la medicament. De exemplu, diferite grade de vascularizare au ca rezultat scăderea fluxului sangvin, a nivelului de oxigen, precum şi diferenţe de pH în fluidele intra‑/extracelulare care pot afecta ionizarea sau activarea medicamentelor şi capacitatea medicamentului de a penetra ţesuturile şi membranele necesare pentru a ajunge la micobacterii.
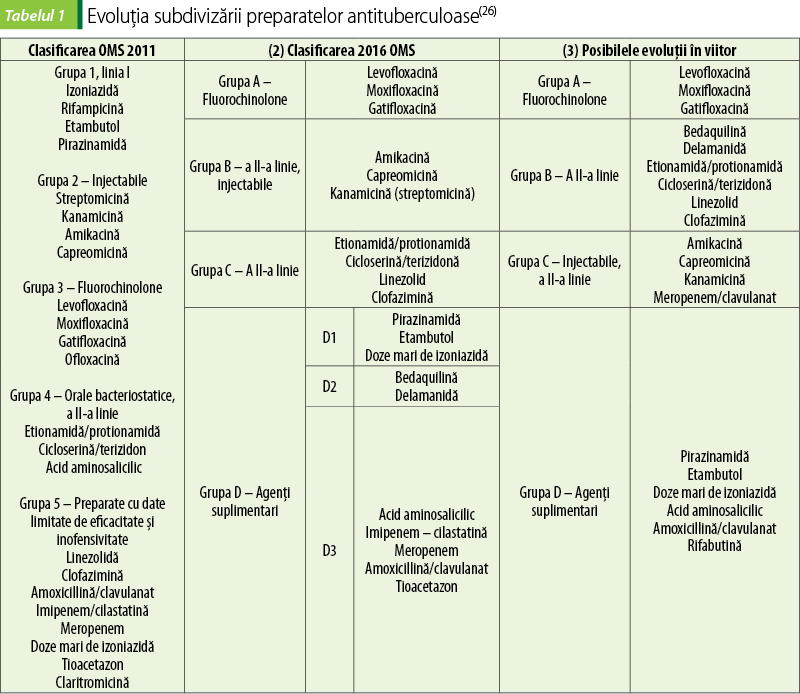
În ultimul deceniu, studiul preparatelor antituberculoase a cunoscut o ascensiune promiţătoare, remarcată şi prin evoluţia locului medicamentelor în tratamentul TBC. Rămâne practic neschimbată poziţia preparatelor utilizate în terapia TBC sensibile la izoniazidă, rifampicină, pirazinamidă şi etambutol, în timp ce în tratamentul formelor cu rezistenţă multiplă (MDR) şi extinsă (XDR) survin modificări semnificative (tabelul 1). Astfel, dacă OMS, în 2011, poziţiona fluorchinolonele pe treapta a III-a, actualmente acestea sunt considerate principala grupă în formele MDR şi XDR ale TBC. O ascensiune similară au avut‑o şi oxazolidinonele (linezolid etc.). Bedacvilina şi delamanida, care au fost incluse în lista de medicamente antituberculoase în 2016 (grupa D2), actualmente sunt preparate de linia a II-a (B) în tratamentul TBC rezistente (tabelul 1).
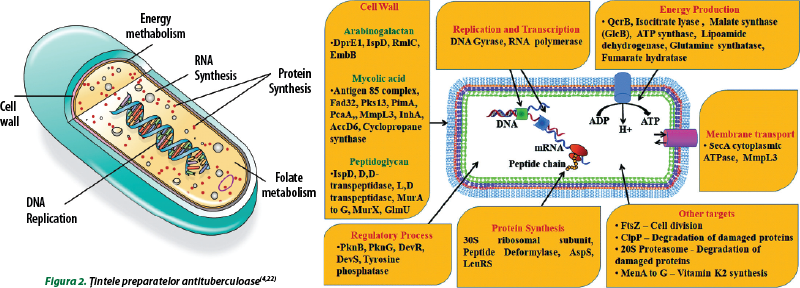
Studiul intens al componentelor structurale şi al ciclului vital al Mycobacterium tuberculosis a permis evidenţierea ţintelor de acţiune ale preparatelor antituberculoase: peretele celular; sinteza proteinelor; sinteza, replicarea şi transcripţia acizilor nucleici; metabolismul energetic etc. (figura 2)(1,4,7,13,16,17,22).
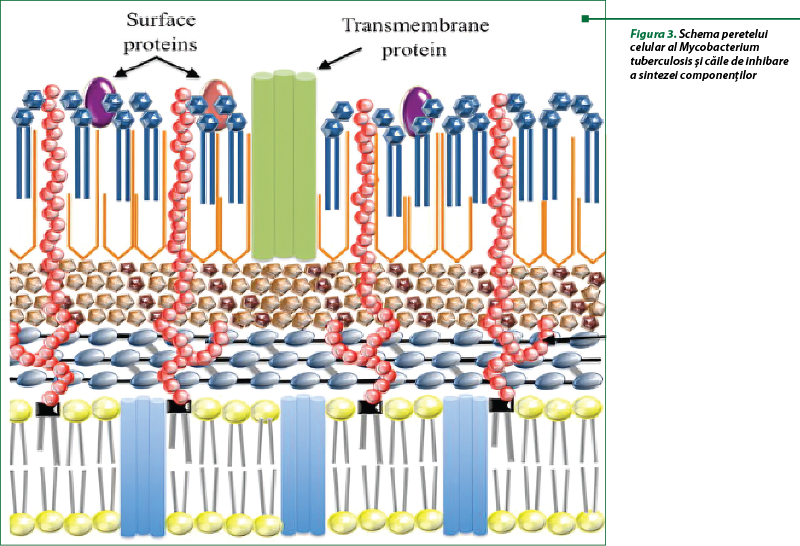
O ţintă importantă pentru elaborarea unor medicamente antituberculoase noi este peretele celular. Odată cu elucidarea componentelor structurale (acizii micolici, arabinogalactanul, peptidoglicanul etc. – figura 3) şi a particularităţilor lor de sinteză şi a enzimelor implicate, s-au specificat următoarele direcţii de acţiune ale preparatelor antituberculoase: inhibitorii sintezei acizilor micolici (izoniazida, etionamida, pretionamida, delamanida, pirazinamida, tioacetazona); inhibitorii sintezei arabinogalactanului (etambutol); inhibitorii sintezei peptidoglicanului (meropenem, imipenem, amoxicilină/clavulanat, cicloserină, capuramicină)(1,7,13,14,16,17,22).
Un component esenţial şi specific pentru micobacteriile tuberculozei sunt acizii micolici, a căror sinteză poate fi dereglată prin: inhibitorii InhA ai enzimei NADH-dependente a enoil-acil-protein reductazei (enoil-ACP) a sintazei acizilor graşi (FASII) – izoniazida, etionamida, pretonamida, delamanida, piridomicina, triclosan; inhibitorii b-cetoacil-ACP sintazei A şi B (KasA/B) – tiolactomicina; inhibitorii b-hidroxiacil-ACP dehidratazei (Had) – NAS-21 şi NAS91; inhibitorii ciclopropan sintazei (CmaA1), esenţială pentru maturarea acizilor micolici – tioacetazona; inhibitorii enzimei Pks13, esenţială la etapa finală de sinteză a acizilor micolici; inhibitorii proteinelor membranare mari (MmpL3), enzimă esenţială în transportul trehalozei monocitrat – adamantil etilendiamine – SQ 109, adamantilureici – AU1235; inhibitorii complexului antigen 85 (Ag85) – proteine cu activitate micoliltransferazică, necesară şi pentru formarea complexului micolic-arabinogalactan-peptidoglican(17,22).
Inhibitori ai InhA
Izoniazida este esenţială în terapia tuberculozei. Obiectivul său molecular este enoil reductaza micobacteriană InhA, care este necesară pentru biosinteza acizilor micolici, componentul dominant al peretelui celular micobacterian lipofil, care este esenţial pentru creştere şi virulenţă. Există două căi distincte de biosinteză a acizilor graşi (FAS). Mamiferele se bazează pe calea FAS I, iar bacteriile – pe FAS II. Micobacteriile conţin ambele căi, cu un subset distinct de enzime incluse în FAS II pentru biosinteza acizilor micolici extraordinar de lungi. Aceste enzime FASII sunt ţinte pentru elaborarea unor medicamente şi un aspect important pentru dezvoltarea unei terapii pentru tulpinile cu polirezistenţă. Izoniazida este un promedicament care necesită activarea prin catalază-peroxidază pentru a inhiba InhA, iar această etapă de bioactivare este locul unde Mycobacterium tuberculosis dezvoltă în principal rezistenţa prin inactivarea acestei enzime neesenţiale. Astfel, elaborarea de substanţe noi sub formă de promedicamente care vizează micobacteriile cu polirezistenţă ar risca să se confrunte cu rezistenţă încrucişată, în cazul în care calea lor de bioactivare este similară cu cea a izoniazidei sau a altor promedicamente utilizate în tratamentul TB, inclusiv etambutol şi pirazinamidă(10,13).
S-au elaborat molecule de plumb cu un nucleu de metil tiazol care sunt inhibitori nanomolari ai InhA cu activitate înaltă faţă de Mycobacterium tuberculosis, un profil de toxicitate preclinic favorabil, cu activitate limitată asupra citocromului CYP450 şi proprietăţi preliminare farmacocinetice favorabile, dar care cedează izoniazidei in vivo. Recent au fost publicate datele unui studiu al derivaţilor de 4-hidroxi-2-piridone care inhibă direct InhA. Compuşii sunt activi la administrarea internă, prezintă o activitate bactericidă puternică împotriva mai multor tulpini cu rezistenţă la izoniazidă, iar in vivo sunt active în modelele cu infecţie acută şi cronică(13).
Piridomicina a fost mai întâi izolată din Streptomyces pyridomyceticus, care este un inhibitor direct al InhA. Preparatul, spre deosebire de izoniazidă şi etambutol, nu necesită bioactivare, se fixează pe situsul activ al InhA şi nu prezintă pericol de rezistenţă încrucişată la aceste medicamente. Medicamentul este capabil să penetreze intracelular şi macrofagele-gazdă, manifestând activitate bactericidă, dar este inactiv împotriva bacteriilor cu replicare lentă, reieşind din modul său de acţiune(13).
Inhibitori ai MmpL3
Proteina membranară micobacteriană (MmpL), care face parte din familia proteinelor de export, este implicată în transportul metaboliţilor din citozolul micobacteriilor. Genomul Mycobacterium tuberculosis conţine 12 gene care exprimă proteinele MmpL, considerate proteine de rezistenţă, care joacă un rol important în supravieţuirea micobacteriilor şi patogeneza tuberculozei. Proteina MmpL3, necesară pentru exportul de acizi micolici sub formă de monomicolaţi de trehaloză în spaţiul periplasmatic sau în membrana exterioară, este esenţială pentru supravieţuirea micobacteriilor, din aceste considerente fiind o ţintă atractivă de compuşi antituberculoşi cu un mecanism nou de acţiune(13,22).
Compusul SQ109, un derivat structural al fragmentului de diamină al etambutolului, s-a dovedit a avea un mecanism diferit de cel al etambutolului – inhibarea MmpL3. Se consideră că SQ109 are proprietăţi polifarmacologice, manifestând activitate pe fungi şi bacterii care nu conţin acizi micolici şi activitate împotriva celulelor latente care nu necesită sinteza activă a peretelui celular. Studiile au relevat că mecanismul de acţiune al SQ109 este cauzat de inhibarea suplimentară a sintezei menachinonei, a respiraţiei celulare şi a sintezei ATP, parţial ca urmare a disipării forţei de tip proton pe membrana citoplasmică. Aceste mecanisme multiple de acţiune antituberculoasă sugerează că SQ109 ar fi un agent eficient pentru terapia tuberculozei polirezistente şi va determina cazuri limitate de rezistenţă(13).
Indolcarboxamidele, o altă clasă de inhibitori de MmpL3, au proprietăţi farmacodinamice excelente, inclusiv activitate bactericidă dependentă de concentraţie şi de timp faţă de un spectru îngust de bacterii Gram‑pozitive şi Gram-negative. Studiile comparative cu SQ109 şi alţi inhibitori ai MmpL3 au demonstrat că indolcarboxamidele se leagă cu diferite situsuri, fapt ce nu dezvoltă rezistenţă încrucişată cu alte medicamente utilizate pentru tratamentul tuberculozei. Compuşii de plumb au o biodisponibilitate orală bună şi prezintă toxicitate limitată. Un avantaj crucial al indolcarboxamidei este acumularea dramatică în plămâni in vivo, cu concentraţii de 5 ori mai mari ca în plasmă, datorită lipofilităţii înalte, fapt firesc pentru rolul MmpL3 în exportul de acizi micolici(13).
O ţintă importantă a devenit arabinogalactanul (figura 4), a cărui sinteză poate fi dereglată prin: 1) inhibitorii DprE1 (decaprenilfosforil-1-ß-ribofuranozei 2-oxidazei) – benzotiazone (macozinom); azaindoli (TBA 7371); 2) inhibitorii arabinosiltransferazei (etambutol, etilendiamine); 3) inhibitorii IspD, enzimă-cheie în calea metileritrizolului fosfat, esenţială în sinteza arabinogalactanului şi PG; 4) inhibitorii RmlC, enzimă-cheie a căii raminozei, esenţială în sinteza arabinogalactanului şi peptidoglicanului (PG)(1,11,18).
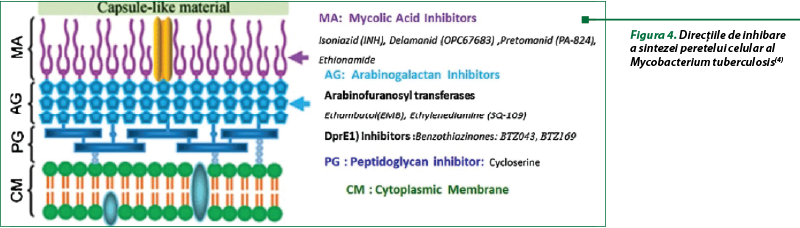
Inhibitorii DprE1
Decaprenilfosforil-p-D-riboza 2’-epimeraza 1 (DprE1), în combinaţie cu DprE2, catalizează epimerizarea decaprenilfosforil-p-D-ribozei la decaprenilfosforil-p-D-arabinoză, asigurând astfel un precursor crucial al polizaharidului de arabinogalactan din peretele celular printr-o serie de oxidare secvenţială. Această enzimă, esenţială pentru supravieţuirea micobacteriilor, a fost identificată ca ţintă pentru benzotiazone, ca potenţiali agenţi antimicobacterieni, cu un mecanism nou de acţiune. Inhibitorii DprE1 sunt consideraţi clasă nouă pentru tratamentul tuberculozei polirezistente, cu rezistenţă încrucişată la opţiunile terapeutice curente(6,10,11,13).
Benzotiazonele au prezentat activitate antimicobacteriană în concentraţie nanomolară în mai multe modele in vivo şi in vitro. Compusul PBTZ 169, moleculă care conţine plumb, este considerat un medicament inovator pentru tuberculoză. Benzotiazonele sunt inhibitori ireversibili, care necesită activarea unei grupări nitro-aromatice pentru activitatea antituberculoasă. Este necesară gruparea nitro, precum şi un grup care îndepărtează metaelectronul, în mod tipic fie un trifluorometil, fie un alt nitro (dinitro benzeni). Gruparea nitro-aromatică este disponibilă pentru o bioreducţie mediată specific micobacteriilor la o grupare nitrozo intermediară, care reacţionează apoi cu un reziduu de cisteină din situsul activ în enzima DprE1, pentru a forma un aduct semimercaptal care inactivează enzima. Este important faptul că metabolismul organismului uman nu are capacitatea de a activa astfel de promedicamente, oferind o fereastră de siguranţă adecvată, şi sunt negative pentru mutageneza ADN(6,13).
O clasă alternativă de inhibitori DprE1 sunt 1,4-azaindolii, derivaţi de imidazo-piridină, cu activitate bactericidă, selectivitate faţă de fosfodiesteraza umană tip 6 (PDE6) şi stabilitate metabolică. Inhibarea PDE6 este în detrimentul acuităţii vizuale, iar moleculele de plumb din aceşti compuşi manifestă o diminuare semnificativă a inhibării acestei enzime şi un profil farmacocinetic bun, menţinând sau îmbunătăţind activitatea antimicobacteriană. Această clasă de inhibitori DprE1 (la 100 mg/kg) a avut o eficacitate comparabilă cu izoniazida într-un model de tuberculoză acută şi cu rifampicina într-un model cronic. O diferenţă importantă pentru această clasă potenţială de medicamente este lipsa rezistenţei încrucişate cu benzotiazonele, deşi au aceeaşi ţintă moleculară, datorită naturii necovalente a inhibării 1,4-azaindolilor(13).
Cea de-a treia clasă de inhibitori DprE1 este reprezentată de compusul TCA1, care împiedică formarea biofilmului micobacterian. Acesta are activitate bactericidă împotriva micobacteriilor sensibile şi cu rezistenţă MDR/XDR, aflate în stare activă şi latentă. TCA1 are un spectru îngust specific de activitate pentru micobacterii şi o activitate bactericidă puternică, sugerând un mecanism nou şi un mare potenţial de opţiune terapeutică. S-a arătat că TCA1 previne formarea enzimei într-o manieră dependentă de doză, printr-o legare competitivă cu enzime ca şi benzotiazonele. O diferenţă importantă este faptul că benzotiazonele nu prezintă activitate pe micobacteriile latente, iar TCA1 reglează expresia genelor asociate cu persistenţa şi toleranţa la medicament. Acest beneficiu suplimentar asupra micobacteriilor latente este considerat a fi datorat unei a doua ţinte a TCA1, o ţintă care nu este esenţială pentru creştere în condiţii optime, dar este relevantă fiziologic atunci când micobacteria se află sub stres antibiotic. În condiţii experimentale s-a demonstrat că un analog TCA1 a influenţat o ţintă secundară, o proteină implicată în biosinteza cofactorului de molibden, iar gena care codifică această proteină este conservată numai în micobacterii, şi nu în alte micobacterii sau specii bacteriene. Cofactorul de molibden este esenţial pentru activitatea nitroreductazei în micobacterii la stresul indus de hipoxie şi oxidul nitric(13).
Cercetările recente au demonstrat că peptidoglicanul este un component al peretelui celular, iar sinteza lui poate fi influenţată prin: 1) inhibitorii D-D-transpeptidazei (amoxicilina/clavulanat); 2) inhibitorii D-D-transpeptidazei şi L-D-transpeptidazei (imipenem, meropenem, faropenem etc.); 3) inhibitori competitivi ai alanin racemazei (Alr) şi D-alanin-D-alanin ligazei (cicloserina); influenţarea translocazei I (capuramicina)(7,9,15,22,25).
Beta-lactamele, efective în tratamentul unei game largi de infecţii bacteriene, nu manifestau activitate antimicobacteriană evidentă. Progresul, referitor la datele noi asupra morfologiei şi ciclului vital al Mycobacterium tuberculosis, a deschis noi posibilităţi de cunoaştere a mecanismelor prin care agenţii b-lactamici pot fi efectivi în tratamentul tuberculozei, inclusiv cu tulpini rezistente. Iniţial s-a contat pe amoxicilină cu acid clavulanic, dar s-a demonstrat o activitate limitată în tratarea infecţiei cu tulpini cu polirezistenţă. Studiile recente au arătat că carbapenemele sunt cele mai potrivite b-lactamine pentru tratarea tuberculozei, deoarece vizează în mod eficient proteinele de legare a penicilinei cu greutate moleculară mare şi inactivează transpeptidazele care formează legăturile 3 → 3, considerate unice pentru peretele celular al Mycobacterium tuberculosis. Concomitent, carbapenemii sunt substraturi mai puţin importante pentru b-lactamază tip BlaC al micobacteriilor, în care este exprimată la niveluri înalte şi inactivează majoritatea altor b-lactame. Meropenemul şi imipenemul în asociere cu acidul clavulanic au demonstrat eficacitate clinică faţă de tulpinile cu polirezistenţă şi rezistenţă extinsă la o administrare de trei ori pe zi prin perfuzie intravenoasă. Avantajul beta-lactaminelor, faţă de alte preparate de a doua linie îl constituie un profil de siguranţă bine definit. Cu toate acestea, complicaţiile la utilizarea acestor antibiotice cu spectru ultralarg în terapia de durată pot să provoace tulburări ale microbiocionozei pacienţilor şi ar putea determina probleme de aderenţă la tratament(13).
Faropenem, un penem cu o stabilitate mai bună sub formă de promedicament valabil pentru administrarea internă, prezintă avantaje pentru tratamentul micobacteriilor cu polirezistenţă. Preparatul are o activitate bactericidă promiţătoare atât faţă de micobacteriile active, cât şi faţă de cele în repaus, active metabolic, similar cu meropenemul. Aceste rezultate confirmă eforturile de a elabora o nouă generaţie de beta-lactamine active faţă de Mycobacterium tuberculosis, cu perspectivele eficacităţii clinice(13,25).
Capuramicina este un antibiotic natural, secretat de Streptomyces griseus, care inhibă biosinteza peptidoglicanului prin direcţionarea translocazei I, o enzimă esenţială şi o ţintă distinctă. Derivaţii de capuramicină, circa 7000 de compuşi, inclusiv produsul SQ641, prezintă potenţă şi selectivitate antimicobacteriană impresionante, o activitate bactericidă rapidă şi un efect postantibiotic de lungă durată, dar solubilitate apoasă scăzută. Pentru a depăşi aceste limitări, s-a elaborat un compus (CPZEN-45) sub formă de aerosol, care se absoarbe eficient de ţesutul pulmonar şi poate crea concentraţii terapeutice relevante la locul primar al infecţiei tuberculoase(13).
Sinteza proteinelor în micobacterii a fost prima ţintă de acţiune a preparatelor antituberculoase (streptomicina), iar ulterior prin elucidarea particularităţilor s-a estimat că această sinteză poate fi dereglată prin: 1) inhibitorii funcţiilor ribozomilor (16S ARNr – streptomocina, amicacina; 30S ARNr – kanamicina, capreomicina; 23S ARNr a 50S: a) oxazolidindinonele: linezolid, sutezolid, delpazolid, contezolid, tedizolid; b) macrolidele: claritromicina; analogii pirazinamidei – pirazinamidă); 2) inhibitorii LeuRS (leucil-ARNt-sintazei) – oxaborolii: GSK-070; 3) inhibitorii AspS (aspratil-ARNt-sintazei) – spiro-oxazolidindione: GSK97G; aminotioazolii: GSK93A; enamide: GSK92A; 4) inhibitorii PDF (peptid deformilazei) – metaloproteină importantă în maturarea polipeptidelor(14,18).
Macrolidele ca antituberculoase
Claritromicina, inhibitor al sintezei proteinelor, a demonstrat eficacitate în infecţia micobacteriană. Din aceste considerente, macrolidele, în baza produsului natural secvenamicina A, sunt intens studiate ca preparate antituberculoase potenţiale. Compusul SEC-9 a demonstrat potenţă micobacteriană, proprietăţi farmacocinetice şi un profil metabolic mai bun (stabilitate înaltă şi inhibare CYP) şi este eficace în modelele murine acute. Produsul SEC-9 are o activitate notabilă împotriva micobacteriilor în faza de replicare, latente şi aflate în intramacrofage, iar în asociere cu alte antituberculoase, îndeosebi bedacvilina şi delamanida, s-a observat o creştere dramatică a activităţii bactericide, cu eliminarea aproape completă a infecţiei cronice la şoareci. O prioritate a macrolidelor ar putea consta în reducerea duratei tratamentului la asocierea cu un regim antituberculos existent(13).
Oxazolidinonele ca antituberculoase
Linezolid, primul derivat de oxazolidinonă, a demonstrat o eficacitate clinică excelentă în tratamentul infecţiilor pulmonare Gram-pozitive rezistente la medicamente. Oxazolidinonele sunt inhibitori ai sintezei proteinelor care se leagă la subunitatea ribozomală 50 a ARN 23S. Utilizarea linezolidului este limitată, până la două săptămâni, deoarece agentul poate provoca mielotoxicitate (ca rezultat al inhibării sintezei proteinelor mitocondriale), citopenie, neuropatii, acidoză lactică şi rabdomioliză. Linezolidul, deşi a demonstrat activitate modestă in vitro şi pe modele de infecţie micobacteriană la şoareci, are o eficacitate robustă în tratarea infecţiilor cu micobacterii multirezistente. Totuşi, investigaţiile în grupul oxazolidinonelor pentru tratamentul tuberculozei cu multirezistenţă au determinat apariţia unor noi analogi, precum tedizolidul, sutezolidul, AZD5847 etc., în vederea evitării sau limitării efectelor toxice(13,18).
Inhibitorii Leucil-tARN sintezei ca antituberculoase – oxaboroele
Leucil-tARN sinteza (LeuRS) este o enzimă importantă pentru sinteza proteinelor şi actualmente a devenit un obiectiv atractiv pentru elaborarea unor inhibitori ai acestei enzime. A fost identificată o clasă de inhibitori care conţin bor, ce acţionează pe locul de editare a LeuRS şi îi întrerupe rolul în sinteza leucil-tARN. Inhibarea acestei ribonucleotide terminale prin ataşarea covalentă a diolilor ribozil la atomul de bor va determina blocarea ARNt cu dereglarea aportului corect de aminoacizi şi prevenirea sintezei proteinelor. Cercetările din această clasă de inhibitori au propus un agent pentru infecţiile fungice şi Gram‑negative, iar actualmente şi de compuşi activi faţă de micobacterii datorită particularităţilor structurale specifice ale LeuRS la Mycobacterium tuberculosis(13).
O ţintă atractivă de acţiune a preparatelor antituberculoase a devenit metabolismului energetic (figura 5), care poate fi influenţat prin: 1) inhibitorii ATP-sintezei (diarilchinoline – bedacvilina, TBAJ-587); 2) inhibitorii lanţului respirator prin Qcrb (complexul citocromului bc1 – imidazopiridine – telacebec, Q203); 3) inhibitorii metabolismului glucidic şi colesterolului (malat sintezei, isocitrat liazei, menachinon sintezei, inhibitorii factorului de creştere operon intracelular); 4) analogii pirazinamidei – pirazinamida(3,5,10,14,27).
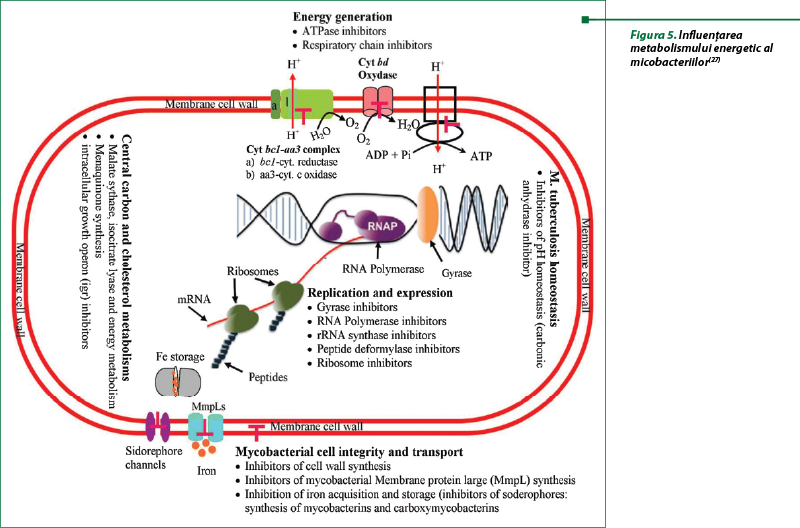
Diarilchinolonele şi antagoniştii lanţului respirator
Diarilchinolonele, bedacvilina şi analogii ei, sunt cele mai avansate medicamente antituberculoase noi aflate în studiu. Bedacvilina manifestă acţiune selectivă şi inhibă specific activitatea ATP-sintezei, esenţială în micobacteriile care se multiplică şi în cele latente, dar nu acţionează asupra celulelor procariote sau eucariote. Micobacteriile rezistente la medicamente sau cele în stare latentă utilizează în calitate de energie ATP, produsă de ATP-sinteză pentru a menţine o membrană energizată. S-a descoperit că inelul rotor al ATP-sintezei se leagă în mod specific la subunitatea c, fapt care a constituit o ţintă pentru noii compuşi antituberculoşi. Acest lucru face bedacvilina un prim preparat pentru acţiune asupra subpopulaţiei latente de micobacterii. Bedacvilina, datorită acestui mecanism de acţiune, distinct de cel al rifampicinei şi izoniazidei, este considerat un component eficient în cazul micobacteriilor cu polirezistenţă. Preparatul manifestă acţiune faţă de tulpinile sensibile şi rezistente la medicamente, cu o concentraţie minimă inhibitoare (CMI) respectiv de 0,03 µg/ml şi 0,12 µg/ml în condiţii experimentale(13).
Eficacitatea mai modestă la om s-a dovedit a fi dependentă de unii factori, precum distribuţia limitată în granulomul uman şi metabolizarea prin intermediul izoenzimei CYP3A4, datorită lipifilităţii înalte. Din aceste considerente sunt posibile interacţiuni farmacocinetice cu inductorii CYP3A4, inclusiv rifampicina, care îi scad activitatea. Utilizarea bedacvilinei a fost asociată cu prelungirea intervalului QT, care impune prudenţă la asocierea cu alte medicamente cu efect similar, inclusiv fluorochinolonele, macrolidele, clofazimina sau cu medicamentele care inhibă CYP3A4. Analiza structurii bedacvilinei a evidenţiat noi posibilităţi de obţinere a unor compuşi cu proprietăţi farmacodinamice şi farmacocinetice care ar permite creşterea eficacităţii şi inofensivităţii(13).
Din diarilchinolone face parte Q203, un compus de imidazopiridină şi care acţionează asupra lanţului respirator, aflat la studiile clinice de fază I. Q203 vizează subunitatea citocromului b a complexului citocrom bc1. Acest complex este o componentă esenţială a lanţului de transport al electronilor şi ai sintezei ATP. Compusul determină o epuizare rapidă a ATP intracelular şi întrerupe homeostazia ATP în micobacteriile latente. Porţiunea de imidazopiridină a fost selectată datorită activităţii sale atât în macrofage, cât şi în mediile libere, cu activitate împotriva izolatelor clinice de micobacterii cu polirezistenţă. Q203 nu inhibă izoformele citocromului P450 şi nu este substrat sau inhibitor al P-glicoproteinei de eflux, ceea ce determină un risc scăzut de interacţiuni medicamentoase. Q203 este un medicament foarte lipofil, cu o legare foarte intensă cu proteinele plasmatice(6,13).
Deşi posibilitatea de influenţare a sintezei acizilor nucleici şi predecesorilor este cunoscută din anii ’60 ai secolului XX (rifampicina, acidul paraaminosalicilic), actualmente s-au elucidat ţinte noi prin: 1) inhibitorii ADN-girazei şi topoizomerazei: fluorchinolonele (levofloxacina, gatifloxacina, moxifloxacina); derivaţii aminobenzimidazolici; derivaţii aminopirazinamidici; novobiocina; nitroimidazoli (metronidazol); flavonoizi (taxofilina – inhibă ADN-giraza şi isoleucil-ARNt-sinteza); 2) inhibitorii ARN-polimerazei (ansamicinele: rifampicina, rifapentina etc.); 3) inhibitorii sintezei ADN (riminofenazine: clofazimina, TBI-166); 4) inhibitorii folaţilor, predecesorii acizilor nucleici (acidul paraaminosalicilic)(18,19).
În ultimii ani, peptidele antimicrobiene le-au trezit un interes cercetătorilor, ca antituberculoase. Peptidele antimicrobiene (PAM) endogene şi exogene (inclusiv de origine entomologică) pot manifesta acţiune directă şi imunomodulatoare. Acţiunea directă se datorează interacţiunii cu învelişul celulei micobacteriene şi în special cu micomembrana şi membrana plasmatică şi cu alte ţinte. Astfel, PAM cationice interacţionează cu suprafaţa anionică a membranei bacteriene, cu formarea complexului peptid-lipidic cu conglomerarea moleculelor peptidice şi alterarea membranei bacteriene. Modificarea respectivă favorizează: a) translocarea peptidelor în toată membrana celulară şi ţinteşte componentele intracelulare: enzimele, acizii nucleici şi alte organele prin difuzarea în citoplasmă; b) formarea porilor, cu dereglarea permeabilităţii membranei; c) inhibarea sintezei peptidoglicanului(2,14).
Acţiunea imunomodulatoare se realizează prin inducerea sintezei citokinelor (interferonilor, interleukinelor). PAM pot fi utilizate ca medicament singur sau în combinaţie cu antibiotice convenţionale. Acestea pot avea efecte faţă de micobacteriile intracelulare care sunt în fagozom, macrofage, granulom sau în plămân. PAM exogene pot avea efecte în faza latentă a tuberculozei, iar în faza acută pot ataca micobacteriile în mod direct(12,21).
În baza analizei prezentate se poate concluziona că preparatele antituberculoase disponibile acţionează prin următoarele mecanisme.
A. Inhibarea sintezei peretelui celular
-
Inhibitorii sintezei acizilor micolici: izoniazida, etionamida, pretionamida, delamanida, pirazinamida, tioacetazona.
-
Inhibitorii sintezei arabinogalactanului: etambutol.
-
Inhibitorii sintezei peptidoglicanului: meropenem, imipenem, amoxicilină/clavulanat, cicloserina, capuramicina.
B. Inhibarea sintezei proteinelor
-
Aminoglicozidele: streptomicina, kanamicina, amicacina.
-
Macrolidele: claritromicina etc.
-
Oxazolidindinonele: linezolid, sutezolid, delpazolid, contezolid etc.
-
Analogii pirazinamidei – pirazinamida.
C. Inhibarea sintezei ARN şi ADN
-
Inhibitorii ADN-girazei şi topoizomerazei: fluorchinolonele – levofloxacina, gatifloxacina, moxifloxacina.
-
Inhibitorii ARN-polimerazei: ansamicinele: rifampicina, rifapentina, rifabutina etc.
-
Inhibitorii sintezei ADN: riminofenazine – clofazimina.
-
Inhibitorii folaţilor, predecesorii acizilor nucleici: acidul paraaminosalicilic.
D. Inhibarea metabolismului energetic
-
Inhibitorii ATP-sintazei: diarilchinoline – bedacvilina.
-
Inhibă metabolismul energetic: pirazinamida.
Aceste date sunt importante în vederea asocierii preparatelor antituberculoase.
Bibliografie
- Abrahams KA, Besra GS. Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology. 2018 Feb; 145(2): 116–133.
- Al Matar M et al. Antimicrobial peptides as an alternative to anti-tuberculosis drugs. Pharmacol Res. 2018 Feb;128:288-305.
- Bei Shi Lee et al. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. J. Biol. Chem. (2019) 294(6) 1936 –1943
- Bhat ZS et al. Drug targets exploited in Mycobacterium tuberculosis: Pitfalls and promises on the horizon. Biomed Pharmacother. 2018 Jul;103:1733-1747.
- Black PA et al. Energy Metabolism and Drug Efflux in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2014, May, 58 ; 5: 2491–2503.
- Brecik M et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol., 2015, 10 (7), pp 1631–1636.
- Catalão MJ, Filipe SR, Pimentel M. Revisiting Anti-tuberculosis Therapeutic Strategies That Target the Peptidoglycan Structure and Synthesis. Front Microbiol. 2019 Feb 11;10:190
- D’Ambrosio L et al. New anti-tuberculosis drugs and regimens: 2015 update. ERJ Open Res. 2015 May; 1(1): 00010-2015.
- Fan YL et al. Fluoroquinolone derivatives and their anti-tubercular activities. Eur J Med Chem. 2018 Feb 25;146:554-563.
- Ferraris DM et al. Mycobacterium tuberculosis Molecular Determinants of Infection, Survival Strategies, and Vulnerable Targets. Pathogens. 2018 Feb 1;7(1). pii: E17.
- Gawad J, Bonde C. Decaprenyl-phosphoryl-ribose 2’-epimerase (DprE1): challenging target for antitubercular drug discovery. Chem Cent J. 2018; 12: 72.
- Gutsmann T. Interaction between antimicrobial peptides and mycobacteria. Biochim Biophys Acta. 2016 May;1858(5):1034-43.
- Hoagland D et al. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv Drug Deliv Rev. 2016 Jul 1; 102: 55–72.
- Khusro A et al. Neoteric advancement in TB drugs and an overview on the anti-tubercular role of peptides through computational approaches. Microbial Pathogenesis. 2018, 114, 80-89.
- Li F et al. In Vitro Activity of β-Lactams in Combination with β-Lactamase Inhibitors against Mycobacterium tuberculosis Clinical Isolates. Biomed Res Int. 2018; 3579832
- Machado D et al. Challenging the Drug-Likeness Dogma for New Drug Discovery in Tuberculosis. Front Microbiol. 2018; 9: 1367.
- North EJ, Jackson M, Lee RE. New approaches to target the mycolic acid biosynthesis pathway for the development of tuberculosis therapeutics. Curr Pharm Des. 2014;20(27):4357-78.
- Olaru ID et al. Novel drugs against tuberculosis: a clinician’s perspective. Eur Respir J. 2015 Apr;45(4):1119-31.
- Pranger AD et al. The Role of Fluoroquinolones in the Treatment of Tuberculosis in 2019. Drugs. 2019; 79(2): 161–171.
- Podany AT, Swindells S. Current strategies to treat tuberculosis. Version 1. F1000Res. 2016; 5: F1000 Faculty Rev-2579.
- Portell-Buj E et al. In vitro activity of 12 antimicrobial peptides against Mycobacterium tuberculosis and Mycobacterium avium clinical isolates. J Med Microbiol. 2019 Feb;68(2):211-215.
- Shaikh NS, Sawarkar SP. Targeting Approaches for Effective Therapeutics of Bone Tuberculosis. J Pharm Microbiol. 2017, 3:1.
- Reiche MA, Warner DF, Mizrahi V. Targeting DNA Replication and Repair for the Development of Novel Therapeutics against Tuberculosis. Front Mol Biosci. 2017 Nov 14;4:75.
- Rihwa Choi MD et al. Recommendations for Optimizing Tuberculosis Treatment: Therapeutic Drug Monitoring, Pharmacogenetics, and Nutritional Status Considerations. Ann Lab Med. 2017 Mar; 37(2): 97–107.
- Sotgiu G et al. Carbapenems to Treat Multidrug and Extensively Drug-Resistant Tuberculosis: A Systematic Review. Int J Mol Sci. 2016 Mar; 17(3): 373.
- Tiberi S et al. Classifying new anti-tuberculosis drugs: rationale and future perspectives. International Journal of Infectious Diseases 2017 V. 56, P. 181-184
- Tuyiringire N et al. Application of metabolomics to drug discovery and understanding the mechanisms of action of medicinal plants with anti-tuberculosis activity. Clin Transl Med. 2018 Oct 1;7(1):29.
- Yong-Soo Kwon. Clinical Implications of New Drugs and Regimens for the Treatment of Drug-resistant Tuberculosis. Chonnam Med J. 2017 May; 53(2): 103–109.
- World Health Organization. Global Tuberculosis Report. 2018, 231p. ISBN 978-92-4-156564-6.
Articole din ediţiile anterioare
FARMACOLOGIE SI FARMACOTERAPIE | Ediţia 1 192 / 2020
Tuberculoza, o boală veche, dar încă prezentă în România
Cristian Daniel Marineci, Cristina Elena Zbârcea, Simona Negreş
Tuberculoza este o infecţie cronică, localizată cel mai adesea la nivelul plămânilor, care se manifestă de obicei după o perioadă de latenţă de la ...
16 martie 2020