Mastocytosis is a rare disease that has been defined as an abnormal accumulation of mast cells in one or more organ systems. Previously considered a myeloproliferative neoplasm, mastocytosis is now classified in its own category under myeloid neoplasms. Since 2016 it was separated into three categories, cutaneous mastocytosis (CM), systemic mastocytosis (SM) and mast cell sarcoma, these diseases occuring in both children and adults. We hereby present a diagnostic algorithm and a short case report of a patient with a rare form of SM.
HEMATO-ONCOLOGY
Mastocitoza – o patologie unică, având multiple faţete
Mastocytosis – unique pathology with multiple facets
Ana Maria Prof. Dr. Vlădăreanu,
Conf. Dr. Horia Bumbea,
Delia Soare,
Alina Mititelu,
Șef de lucrări dr. Minodora Onisâi
First published: 27 martie 2019
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.46.1.2019.2314
Abstract
Rezumat
Mastocitoza este o boală rară care a fost definită ca o acumulare anormală de mastocite în unul sau mai multe sisteme de organe. Anterior considerată o neoplazie mieloproliferativă, mastocitoza este acum clasificată ca făcând parte din neoplasmele mieloide. Din 2016 a fost separată în trei categorii: mastocitoza cutanată (CM), mastocitoza sistemică (SM) şi sarcomul cu celule mastocitare, aceste boli apărând atât la copii, cât şi la adulţi. Prezentăm un algoritm de diagnosticare şi un scurt raport de caz al unui pacient cu o formă rară de SM.
Mastocytosis refers to a group of disorders characterized by excessive mast cell accumulation in one or more tissues. Mastocytosis is subdivided into two groups of disorders:
-
Cutaneous mastocytosis (CM) describes forms of mastocytosis that are limited to the skin.
-
Systemic mastocytosis (SM) describes forms of mastocytosis in which pathologic mast cells infiltrate multiple extracutaneous organs – including the bone marrow (BM), spleen, and the gastrointestinal (GI) tract, with or without skin involvement.
Cutaneous mastocytosis (CM) is most frequently seen in children and may regress or even disappear in adulthood. The cutaneous form of the disease is divided into maculopapular CM (also termed urticaria pigmentosa), diffuse cutaneous mastocytosis, and localized mastocytoma of skin(1).
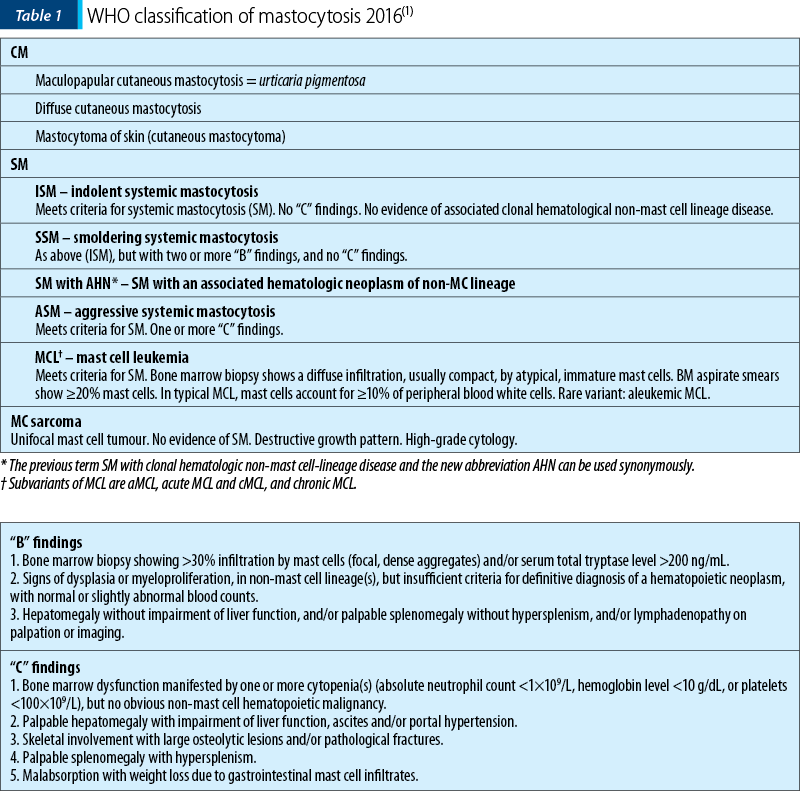
Systemic mast cell disorders in most instances appear to be clonal disorders of the mast cell and its progenitors. Systemic mastocytosis (SM) is a chronic, persistent disease. A somatic c-kit mutation at codon 816 is often detectable in hematopoietic cells. The clinical course of mastocytosis is variable, ranging from indolent to aggressive. Five categories of the disease are recognized in the updated World Health Organization (WHO) classification. SM is divided into ISM (indolent systemic mastocytosis), smoldering SM (SSM), SM with an associated hematologic (non-MC lineage) neoplasm (SM-AHN), ASM (aggressive systemic mastocytosis), and MCL (mast cell leukemia). AHN is a novel abbreviation and can be used instead of (or synonymously to) the previous term “associated hematologic non-mast cell-lineage disease” (SM-AHNMD)(2).
The pathognomonic feature in SM is the multifocal infiltration of MCs in various internal organs, including the BM. The criteria for the diagnosis of systemic mastocytosis are listed in Table 2(3).

SM criteria were defined by WHO in 2001 and have been confirmed in the 2008 and 2016 WHO updates.
The diagnosis of SM is to be established if the major and at least one minor SM criterion or three minor SM criteria have been detected.
In a significant subgroup of patients (10-35%), an associated clonal hematologic non-mast cell lineage disorder occurs. These AHNs can be classified according to recently established WHO criteria. Most AHNs resemble myeloid malignancies such as acute myeloid leukemia, myeloproliferative disorders or myelodysplastic syndromes. In only a few cases, lymphoproliferative disorders are diagnosed(4).
Patients with SM-AHN have a less favorable prognosis regarding survival when compared to indolent SM.
No general guidelines for the treatment of patients with SM-AHN have been established so far; additional genetic abnormalities have been reported. A reasonable straightforward approach may be to treat the AHN in those patients in the same way as if no coexisting SM existed. Patients with cutaneous or indolent systemic disease are treated symptomatically. Patients with aggressive disease are candidates for cytoreductive therapy. The use of ‘Kit-targeting’ tyrosine kinase inhibitors are best selected following a mutational analysis of c-kit. For instance, the D816V mutation appears to be associated with relative resistance to imatinib. However, imatinib has been used with success in patients with SM-hypereosinophilic syndrome (HES) and the FIP1L1/PDGFRA fusion gene, and in a patient with mastocytosis with a mutation outside of codon 816. The value of bone marrow transplantation remains under investigation.
The association of systemic mastocytosis (SM) with a chronic myeloproliferative neoplasm is the second category by frequency. Published descriptions of the clinical and pathologic features of SM with AHN are largely limited to individual case reports.
Case presentation
We present the case of a 34-year-old woman who presented in our clinic with thrombocytosis, and mild splenomegaly and hepatomegaly; she also had failure of in vitro fertilization. The clinical suspicion was of chronic myeloproliferative neoplasm (MPN).
Bone marrow biopsy and molecular examinations (BCR-ABL, JAK2 V617F, MPL, CALR) were performed. Bone marrow trephine biopsy examination (histology and immunohistochemistry for CD25 and tryptase) revealed a hypercellular bone marrow with 16-18% mastocytes disposed focally and perivascularly and groups of >15 mast cells, some of them spindle-shaped, and frequent groups of megakaryocytes, polymorphic, most of them gigantic megakaryocytes, with hyperlobulated nuclei; the aspect was compatible with SM associated with MPN, essential thrombocythemia (ET) type. The JAK2 V617F mutation, which is associated with MPNs, was detected, with an allele burden of 12%, but KIT/Asp816Val, which is reported in approximately 80% of SM, was absent, and the serum tryptase basal level was 25.5 µg/L. We established the diagnosis of systemic mastocytosis associated with essential thrombocytemia – SM with AHN/SM with an associated hematologic neoplasm of non-MC lineage, as she fulfilled the major criterion and one minor criterion for SM.
The treatment was based on continuous mediator therapy anti-H1 and anti-H2 receptor, antiplatelet (Aspenter® 75 mg/day) and cytoreductive therapy (Anagrelid® 0.5 mg – adjusted dose at the level of thrombocytes). She tolerated the therapy very well, the platelet count was normalized, and the clinical picture was significantly improved.
Awareness on mastocytosis
In Romania, we celebrate on March 25th the national day dedicated to raising awareness on mastocytosis. With this occasion, every year, the Bucharest University Emergency Hospital, in collaboration with the Romanian Mastocytosis Support Association, organizes the National Mastocytosis Conference, which consists in medical presentations, panel discussions and support group sessions. These conferences provide a unique supportive environment in which patients, caregivers, and families learn to self-advocate, and are offered access to research and clinical trial information. Patients, caregivers, and physicians can interact outside of the medical environment to promote more positive relationships in a casual atmosphere.
At the venues, there were present: Sorina Pintea – the Ministry of Health; dr. Cristian Grasu – Ministry of Health Secretary of State; dr. Adriana Nica – the manager of the Bucharest University Emergency Hospital; prof. dr. Ana-Maria Vlădăreanu – the Head of the Hematology Clinic; assoc. prof. dr. Horia Bumbea; assoc. prof. dr. Cora Pop; assoc. prof. dr. Lucian Negreanu; assoc. prof. dr. Sabina Zurac; assoc. prof. dr. Camelia Dobrea; dr. Camelia Berghea; dr. Poliana Leru; Dorica Dan – president of ANBR, and Nicoleta Vaia – the President of Romanian Mastocytosis Support Association.
The topics discussed at the conferences varied from mastocytosis in pediatric population to systemic mastocytosis in adult patients, as well as the implications of mastocytosis in allergology and the difficulties of the morphological diagnosis.
At the Bucharest University Emergency Hospital, there is a Center of Excellence in Mastocytosis. The center is an active member of the ECNM (The European Competence Network on Mastocytosis), and performs a multidisciplinary approach with the help of specialists from allergology, dermatology, hematology and pathology. We currently treat over 50 patients, of which 21 were diagnosed in 2018, the year our Clinic of Hematology was approved as a National Center of Excellence in Rare Diseases. In Romania, the number of the diagnosed patients with mast cell disorders is 252, of which 180 adults and 73 pediatric patients.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017; 129(11):1420–1427. doi:10.1182/blood-2016-09-731893.
- Valent P, Akin C, Hartmann K, et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Future. Cancer Res. 2017; 77(6):1261–1270. doi:10.1158/0008-5472.CAN-16-2234.
- Horny HP, Valent P. Histopathological and Immunohistochemical Aspects of Mastocytosis. International Archives of Allergy and Immunology. 2002; 127. 115-7. 10.1159/000048180.
- Sperr RW, Horny HP, Valent P. Spectrum of Associated Clonal Hematologic Non-Mast Cell Lineage Disorders Occurring in Patients with Systemic Mastocytosis. International Archives of Allergy and Immunology. 2002; 127. 140-2. 10.1159/000048186.
- Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nuñez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001 Jul; 25(7):603-25.
Articole din ediţiile anterioare
CANCERUL DE CANAL ANAL | Ediţia 1 / 2016
Cancerul de canal anal - aspecte legate de diagnostic și tratament
Laurenţiu Simion, Alexandru Grigorescu, Angela Mădălina Lazăr, Şerban Marinescu
Cancerul anal reprezintă o neoplazie rară, constituind aproximativ 1,5% din totalitatea cancerelor digestive, dar rămâne o preocupare deosebită din...
24 martie 2017
REVIEW | Ediţia 1 62 / 2023
Decision making in Merkel cell carcinoma – surgical oncologist point of view
Claudiu Daha, Eugen Brătucu, Hortensia Moisă, Virgiliu Mihail Prunoiu, Laurenţiu Simion
Carcinomul cu celule Merkel (MCC) nu este atât de bine cunoscut precum alte tumori maligne ale pielii, deşi s-a constatat ca are o rată mai crescut...
23 martie 2023
ORIGINAL STUDY | Ediţia 3 / 2016
Studiul erorilor diagnostice și terapeutice corelate cu studiul markerilor tumorali cu impact în prognosticul pacienților cu cancer localizat la cap și gât
Ioan Andrei, Anda Crişan, Florinel Bădulescu, Michael Schenker
Introducere. Alegerea protocolului de tratament pentru pacienții cu carcinoame ale sferei ORL stadii avansate locoregional, rămâne o discuție desc...
07 martie 2017
HEMATO-ONCOLOGY | Ediţia 1 50 / 2020
Limfomul nodal de zonă marginală – etiopatologie, diagnostic şi tratament
Lidia Felicia Mihai
Limfomul nodal de zonă marginală este un tip rar de limfom non-Hodgkin indolent. Acesta afectează în principal ganglionii limfatici, dar prezintă...
30 martie 2020