Familial chylomicronemia syndrome represents a rare disease affecting the lipoprotein metabolism, through decreased levels of lipoprotein lipase (LPL) or apolipoprotein C-II, characterized also by elevated levels of circulating triglycerides. The clinical features of the disease are represented by the lipemic appearance of the patient’s serum (secondary to the high levels of circulating chylomicrons and very low density lipoproteins), splenomegaly, abdominal pain – acute pancreatitis, lipemia retinalis and xanthomas. The authors present the case of a 38-day-old male infant, with no previous history of other diseases, who was diagnosed with familial chylomicronemia after the medical staff observed – during blood sampling for another reason – the lipemic (milky) appearance of the serum. The lipid profile analysis revealed a marked elevation of triglycerides (over the 99th percentile), supporting the diagnosis of a defect in the synthesis/metabolism of lipoproteins. Circulating lipoprotein electrophoresis after 12 hours of fasting demonstrated high levels of chylomicrons. Using a specific diet – medium-chain triglycerides milk formula, resulted in rapid improvement of the serum lipid profile during follow-ups.
Chilomicronemia familială – cauză rară de dislipidemie la sugar
Familial chylomicronemia – a rare cause of dyslipidemia in newborn
First published: 28 decembrie 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.52.4.2018.2158
Abstract
Rezumat
Chilomicronemia familială este o afecţiune rară a metabolismului lipoproteinelor, determinată de deficitul de lipoprotein-lipază (LPL)/apolipoproteina C-II şi caracterizată prin creşterea valorii trigliceridelor serice. Din punct de vedere clinic, pacienţii prezintă plasmă intens lipemică (din cauza prezenţei în cantităţi mari a chilomicronilor şi a lipoproteinelor cu densitate foarte mică – VLDL), splenomegalie, dureri abdominale – pancreatită acută, lipemia retinalis şi xantoame. Autorii prezintă cazul unui sugar în vârstă de 38 de zile, fără antecedente personale patologice semnificative, diagnosticat cu această afecţiune după ce s-a observat întâmplător – în urma recoltării de ser în cursul unei spitalizări – că plasma sa avea aspect intens lipemic (milky). Analizarea ulterioară a profilului lipidic a arătat creşterea marcată (peste percentila 99) a trigliceridelor serice, ridicându-se astfel suspiciunea existenţei unui defect de sinteză/metabolizare al lipoproteinelor. Electroforeza lipoproteinelor serice, efectuată din ser obţinut după 12 ore de post, a demonstrat prezenţa chilomicronilor într-un procent semnificativ crescut. Tratamentul igieno-dietetic, respectiv alimentarea sugarului cu formulă de lapte având ca sursă principală de lipide trigliceridele cu lanţ mediu (MCT), a avut ca efect îmbunătăţirea marcată a profilului lipidic la controalele succesive efectuate.
Introducere
Chilomicronemia – cunoscută anterior sub denumirea de hiperlipoproteinemia de tip I – reprezintă o patologie severă, caracterizată prin acumularea de chilomicroni în plasma pacientului – à jeun(1). Această afecţiune rară a metablismului lipoproteinelor este determinată de deficitul de lipoprotein-lipază (LPL) sau de cel al apolipoproteinei C-II, consecinţa fiind creşterea dramatică a valorii trigliceridelor serice.
Din punct de vedere clinic, pacienţii pot prezenta xantoame cutanate, hepatosplenomegalie, pancreatită acută, iar în ceea ce priveşte aspectul serului în momentul recoltării – acesta este intens lipemic, ca urmare a prezenţei în cantităţi mari a chilomicronilor şi a trigliceridelor(2). Când valoarea trigliceridelor depăşeşte 4000 mg/dl, arteriolele şi venulele de la nivelul retinei capătă o culoare rozată – fenomen întâlnit sub denumirea de lipemia retinalis. Această coloraţie este reversibilă şi nu va afecta pe termen lung vederea pacienţilor(3). Este de subliniat faptul că cea mai severă manifestare clinică întâlnită în cadrul chilomicronemiei este pancreatita acută, aceasta fiind asociată cu o mortalitate de 5-6%, cel mai frecvent în urma necrozei pancreatice(4).
Cel mai important pas în managementul acestor pacienţi este reprezentat de regimul igieno-dietetic. Se va avea în vedere restricţia aportului lipidic la aproximativ 15% din necesarul energetic zilnic, iar sugarii vor primi lapte îmbogăţit cu trigliceride cu lanţ mediu (MCT). Aceste trigliceride nu sunt încorporate în chilomicroni, fiind absorbite direct în circulaţia portală(5). Cu toate acestea, în cazul hipertrigliceridemiilor foarte accentuate, tratamentul igieno-dietetic poate fi insuficient pentru a preveni pancreatita, astfel de situaţii impunând iniţierea unui tratament nefarmacologic, cum ar fi plasma exchange(6).
Prezentare de caz
N.S. este un pacient de sex masculin, care la vârsta de 38 de zile a fost internat la Spitalul de Urgenţă pentru Copii „M.S. Curie” pentru următoarea simptomatologie: febră (maximum 39°C) şi tuse spastică – debutată în urmă cu aproximativ 24 de ore. Din antecedentele personale fiziologice reţinem faptul că este primul copil al cuplului, extras prin operaţie cezariană la vârsta gestaţională de 39 de săptămâni, având o greutate la naştere de 3040 de grame, scor Apgar 10, alimentat exclusiv cu lapte matern. Antecedentele personale patologice erau nesemnificative, iar dintre antecedentele heredocolaterale menţionăm faptul că tatăl a fost diagnosticat cu dislipidemie de către medicul de familie, fără a urma până la momentul respectiv vreun tratament.
Din punct de vedere clinic, în momentul internării pacientul avea greutate de 4100 de grame, prezenta stare generală medie, era febril (38,4°C), tegumentele erau palide, fără erupţii, fără edeme. La examinarea aparatului respirator s-a observat obstrucţie nazală importantă, tuse spastică în accese, saturaţia în aerul atmosferic era O2 = 96%, fără să prezinte sindrom funcţional respirator. Era echilibrat din punct de vedere cardiohemodinamic şi digestiv, fără hepatosplenomegalie, diureza era prezentă, fontanela anterioară normotensivă. Din punct de vedere neurologic, sugarul era dezvoltat corespunzător vârstei.
În urma recoltării serului în vederea efectuării bilanţului biologic al pacientului s-a observat că acesta avea un aspect intens lactescent/lipemic (figurile 1, 2 şi 3). Profilul lipidic efectuat ulterior, după 12 ore de post, a evidenţiat o creştere impresionantă a valorilor lipidelor totale (5595 mg/dl), hipertrigliceridemie severă (1761 mg/dl) şi hipercolesterolemie moderată (colesterol total 420 mg/dl, LDLc=1314 mg/dl, VLDLc=352 mg/dl). Astfel, s-a ridicat suspiciunea existenţei unei afecţiuni metabolice, respectiv un deficit de sinteză şi metabolism al lipoproteinelor. În continuarea demersului diagnostic s-a efectuat electroforeza lipoproteinelor serice şi s-a observat prezenţa unui procent crescut de chilomicroni – 12,7% (valori normale <0,2%), însoţit de un procent scăzut al alfa- şi, respectiv, beta-lipoproteinelor (19,8%, respectiv 27,4%). Astfel, diagnosticul final al sugarului a fost cel de chilomicronemie familială.

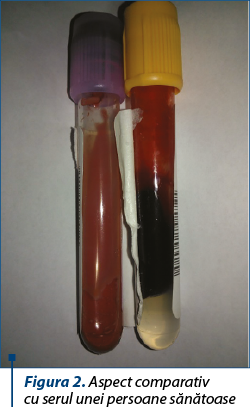
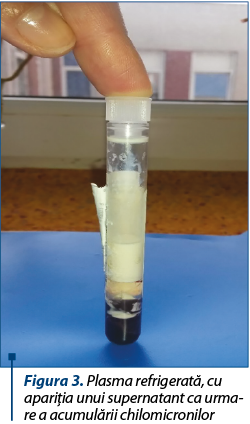
În ceea ce priveşte abordarea terapeutică a pacientului, aceasta a constat în modificarea alimentaţiei, respectiv oprirea alimentaţiei la sân şi înlocuirea cu o formulă de lapte îmbogăţită cu MCT-uri. După aproximativ două luni de la introducerea dietei, profilul lipidic al pacientului s-a îmbunătăţit substanţial – valoarea lipidelor totale a scăzut până la valoarea de 798 mg/dl, a trigliceridelor până la 346 mg/dl şi a LDLc până la 95 mg/dl.
Discuţii
În acest studiu am prezentat cazul unui sugar al cărui ser era intens lipemic şi care a fost diagnosticat ulterior cu o afecţiune metabolică, respectiv chilomicronemie familială. Caracteristica acestei afecţiuni a metabolismului lipidic este reprezentată de existenţa valorii extrem de crescute a trigliceridelor serice (în general, peste percentila 99).
În ceea ce priveşte hipertrigliceridemiile, acestea pot fi secundare unor afecţiuni sistemice – diabet decompensat, hipotiroidism, sindrom nefrotic – sau secundare obezităţii, abuzului de alcool ori administrării unei varietăţi de medicamente(7). Odată excluse aceste etiologii, medicul curant ar trebui să suspicioneze existenţa unei patologii genetice, cum ar fi chilomicronemia familială.
Chilomicronemia familială reprezintă o afecţiune autozomal recesivă, determinată cel mai frecvent de deficitul de lipoprotein-lipază. Aceasta este sintetizată în miocite şi adipocite şi joacă un rol important în multiple etape din metabolismul lipidelor, inclusiv în hidroliza chilomicronilor şi a lipoproteinelor cu densitate foarte mică. Multiple mutaţii genetice au fost asociate cu această patologie – mutaţia APOA5 (cofactor al lipoprotein lipazei), mutaţia LMF1 (factor de maturizare al lipazei 1) sau mutaţia GPIHBP1 (transportă lipoprotein-lipaza până la nivelul lumenului capilar)(8), existând astfel posibilitatea stabilirii unui diagnostic genetic, de certitudine. În cazul pacientului nostru, aceste testări nu au putut fi efectuate din motive logistice, electroforeza lipoproteinelor serice fiind cea care a susţinut în final diagnosticul.
În general, debutul clinic al chilomicronemiei este în copilărie, însă în aproximativ 25% din cazuri acesta se petrece în perioada de sugar(9). Iar cel mai dramatic tablou clinic este cel al pancreatitei acute, această afecţiune fiind responsabilă de aproximativ 7% din totalul pancreatitelor întâlnite în populaţia pediatrică. Boala apare rar dacă nivelul trigliceridelor serice nu depăşeşte 1760 mg/dl, însă reprezintă o urgenţă medico-chirurgicală, iar managementul dislipidemiei este de o importanţă extremă în prevenţia acestei complicaţii posibil fatale(10-12).
Restricţia lipidică, cu administrarea de formule care conţin trigliceride cu lanţ mediu, reprezintă prima linie de tratament. Şi, cu toate că asocierea dietei cu agenţi hipolipemianţi (fibraţi), cu Omega-3 şi/sau orlistat, poate scădea valoarea nivelului trigliceridelor serice, rezultatele rămân controversate(13). Totuşi, Becker et al. au concluzionat că administrarea de fibraţi reprezintă o alternativă sigură şi eficientă pentru a trata hipercolesterolemia în populaţia pediatrică(14).
O altă abordare terapeutică este plasmafereza, aceasta fiind una dintre modalităţile de tratament al hipertrigliceridemiilor, menţionată în ghidul 2010 al American Society of Apheresis(15). Stefanuti et al. au raportat cazul unui sugar în vârstă de 3 luni, diagnosticat cu chilomicronemie şi care a fost supus procedeului de plasmafereză, în vederea evitării apariţiei pancreatitei acute. Rezultatele obţinute au fost spectaculoase, valoarea triglicerdelor serice scăzând de la 167 mmol/L la 11,68 mmol/L în aproximativ 72 de ore de la efectuarea plasmaferezei(16). Cu toate acestea, mici studii clinice care au comparat managementul conservator cu plasmafereza au concluzionat că cea din urmă nu prezintă beneficii semnificative(17).
Recent şi-a făcut apariţia o nouă modalitate de tratament cu lomitapide. Această moleculă este aprobată în America de Nord şi în Europa în vederea tratamentului hipercolesterolemiei familiale homozigote, demonstrându-şi eficienţa prin scăderea valorii trigliceridelor serice cu până la 30-40%(18).
Concluzii
Chilomicronemia familială este în general o afecţiune întâlnită în copilărie şi în adolescenţă. Cu toate acestea, există situaţii în care este descoperită în perioada de sugar – cel mai frecvent întâmplător –, ca urmare a recoltării de ser pentru o altă patologie. Diagnosticarea cât mai precoce şi intervenţia terapeutică cât mai timpurie, respectiv modificarea dietei şi administrarea de agenţi hipolipemianţi, pot îmbunătăţi prognosticul pacienţilor, scăzând semnificativ riscul de a dezvolta complicaţia cea mai redutabilă a acestei patologii – pancreatita acută.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Brahm AJ, Hegele RA. Chylomicroaemia: current diagnosis and future therapies. Nat Rev Endocrinol. 2015; 11:352-362
- Guder WG, Narayanan S, Wisser H, Zawta B. Diagnostic Samples: From the Patient to the Laboratory: The Impact of Preanalytical Variables on the Quality of Laboratory Results. Wiley, New York, NY, USA, 4th edition, 2009
- Burnett JR, Hooper AJ, Hegele RA. Familial Lipoprotein Lipase Deficiency. 1999 Oct 12 [Updated 2017 Jun 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018
- Valdivielso P, Ramírez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med. 2014;25:689–694.
- Gotoda T, Shirai K, Ohta T, Kobayashi J, Yokoyama S, Oikawa S, et al. Diagnosis and management of type I and type V hyperlipoproteinemia. J Atheroscler Thromb. 2012;19:1–12.
- Jung MK, Jin J, Kim HO, et al. A 1-month-old infant with chylomicronemia due to GPIHBP1 gene mutation treated by plasmapheresis. Ann Pediatr Endocrinol Metab. 2017;22(1):68-71.
- Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. 2007;176:1113–1120.
- Davies BS, Beigneux AP, Fong LG, Young SG. New wrinkles in lipoprotein lipase biology. Curr Opin Lipidol. 2012;23:35-42
- Sugandhan S, Khandpur S, Sharma VK. Familial chylomicronemia syndrome. Pediatric Dermatology. 2007; vol. 24, no. 3, pp. 323–325.
- Gan SI, Edwards AL, Symonds CJ, Beck PL. Hypertriglyceridemia-induced pancreatitis: a case-based review. World Journal of Gastroenterology. 2006; vol. 12, no. 44, pp. 7197–7202.
- Fortson MR, Freedman SN, Webster III PD. Clinical assessment of hyperlipidemic pancreatitis. American Journal of Gastroenterology. 1995; vol. 90, no. 12, pp. 2134–2139.
- Searles GE, Ooi TC. Underrecognition of chylomicronemia as a cause of acute pancreatitis. Canadian Medical Association Journal. 1992; vol. 147, no. 12, pp. 1806–1808.
- Mohandas MK, Jemila J, Ajith Krishnan AS, George TT. Familial chylomicronemia syndrome. Indian J Pediatr. 2005;72:181.
- Becker M, Staab D, Von Bergman K. Long-term treatment of severe familial hypercholesterolemia in children: effect of sitosterol and bezafibrate. Pediatrics. 1992;89:138-42.
- Szczepiorkowski ZM, Winters JL, Bandarenko N, Kim HC, Linenberger ML, Marques MB, et al. Guidelines on the use of therapeutic apheresis in clinical practice – evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010;25:83-177.
- Stefanutti C, Gozzer M, Pisciotta L, D’Eufemia P, Bosco G, Morozzi C, et al. A three month-old infant with severe hyperchylomicronemia: molecular diagnosis and extracorporeal treatment. Atheroscler Suppl. 2013;14:73-6.
- Valdivielso P, Ramírez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med. 2014;25:689-94.
- Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide. JAMA Intern Med. 2014;174:443-7.