Oro-facial clefts are the most common birth defects of the cephalic region and the second frequency malformation in newborns, involving a variety of abnormalities in the upper lip and upper jaw. In 10-30% of cases occurs only cleft lip, the cleft palate occurs in only about 25% and 40-50% of cases are complete labio-maxillo-palatine cletfs, which may be unilateral or bilateral. The malformation is invalidated by its consequences on multiple levels: aesthetic, psychological, speech, nutrition, of malformations associated, therefore the therapeutic management begins with antenatal diagnosis and continues immediately after birth, with a complex team of geneticist, pediatrician, pediatric surgeon, plastic surgeon, oro-maxillo-facial doctor, dentist, orthodontist, speech, ear, nose and throat doctor, psychotherapist. Long term outcome is favorable if the patient is treated by a multidisciplinary team that will follow him up into adulthood, facilitating access to all stages of medical, surgical and psychological counseling needed to ensure the integration into society and self-esteem.
Despicăturile labio-maxilo-palatine
Orofacial clefts
First published: 09 noiembrie 2016
Editorial Group: MEDICHUB MEDIA
Abstract
Rezumat
Despicăturile labio-maxilo-palatine sunt cele mai frecvente malformaţii congenitale ale regiunii cefalice şi a doua malformaţie congenitală ca frecvenţă la nou-născuţii vii, implicând o diversitate de anomalii la nivelul buzei superioare şi maxilarului superior. În 10-30% din cazuri apare doar diviziunea buzei, în aproximativ 25% doar diviziunea palatului şi 40-50% din cazuri au diviziune completă, labio-maxilo-palatină, care poate fi uni- sau bilaterală. Malformaţia este invalidantă prin consecinţele ei pe multiple planuri: estetic, psihologic, al vorbirii, al nutriţiei, al malformaţiilor asociate, de aceea managementul terapeutic al acestei afecţiuni începe cu diagnosticul antenatal şi continuă imediat postnatal, într-o echipă alcătuită din genetician, pediatru, chirurg pediatru, chirurg plastician, medic oro-maxilo-facial, stomatolog, ortodontist, logoped, medic otorinolaringolog, psihoterapeut. Evoluţia pe termen lung este favorabilă în cazul preluării pacientului de către echipa multidisciplinară care îl va urmări până la vârsta adultă, asigurându-i accesul la toate stadiile tratamentului medical, chirurgical, precum şi consilierea psihologică necesară integrării în societate şi asigurării respectului de sine.
Despicăturile labio-maxilo-palatine sunt cele mai frecvente malformaţii congenitale ale regiunii cefalice şi implică o diversitate de anomalii la nivelul buzei superioare şi maxilarului superior, uneori şi a nasului şi a aparatului auditiv, implicând o lipsă de substanţă uni- sau bilaterală la nivelul zonei afectate. Despicăturile apar cu o frecvenţă ridicată, de 0,8-2,7/1000 nou-născuţi, cu uşoare variaţii în funcţie de regiunea geografică şi rasă, fiind, după unele studii de specialitate, a doua malformaţie congenitală ca frecvenţă la nou-născuţii vii(1). Cauzele apariţiei acestor malformaţii sunt plurifactoriale, fiind implicaţi atât factori genetici, afecţiuni materne (diabet zaharat, obezitate, deficit de acid folic), cât şi factori de mediu (medicamente, substanţe chimice, fumat, consum de alcool, infecţii virale)(2,3).
În unele cazuri familiale au fost identificate mutaţii ale genei MSX1 (respectiv la 2% dintre pacienţi, care asociază şi anomalii dentare, cu transmitere autozomal-dominantă şi cu risc de recurenţă de 50%), sau mutaţii ale genei TBX22, cu transmitere X-linkată. Alte gene posibil implicate în apariţia despicăturilor labiale sau palatine sunt VAX1, FGFR2, BMP4 și IRF6(3). Pentru cazurile izolate, riscul de recurenţă variază între 2% şi 10%.
Deşi cauzele apariţiei acestor malformaţii nu sunt încă pe deplin cunoscute, cert este faptul că în timpul embriogenezei, între a 15-a şi a 60-a zi de la fecundare, apar anomalii de fuziune ale celor cinci muguri faciali (mugurele fronto-nazal, doi muguri maxilari şi doi muguri mandibulari), cu expresii fenotipice variate. În 10-30% din cazuri apare doar diviziunea buzei, în aproximativ 25% doar diviziunea palatului şi 40-50% din cazuri au diviziune completă, labio-maxilo-palatină, care poate fi uni- sau bilaterală(4).
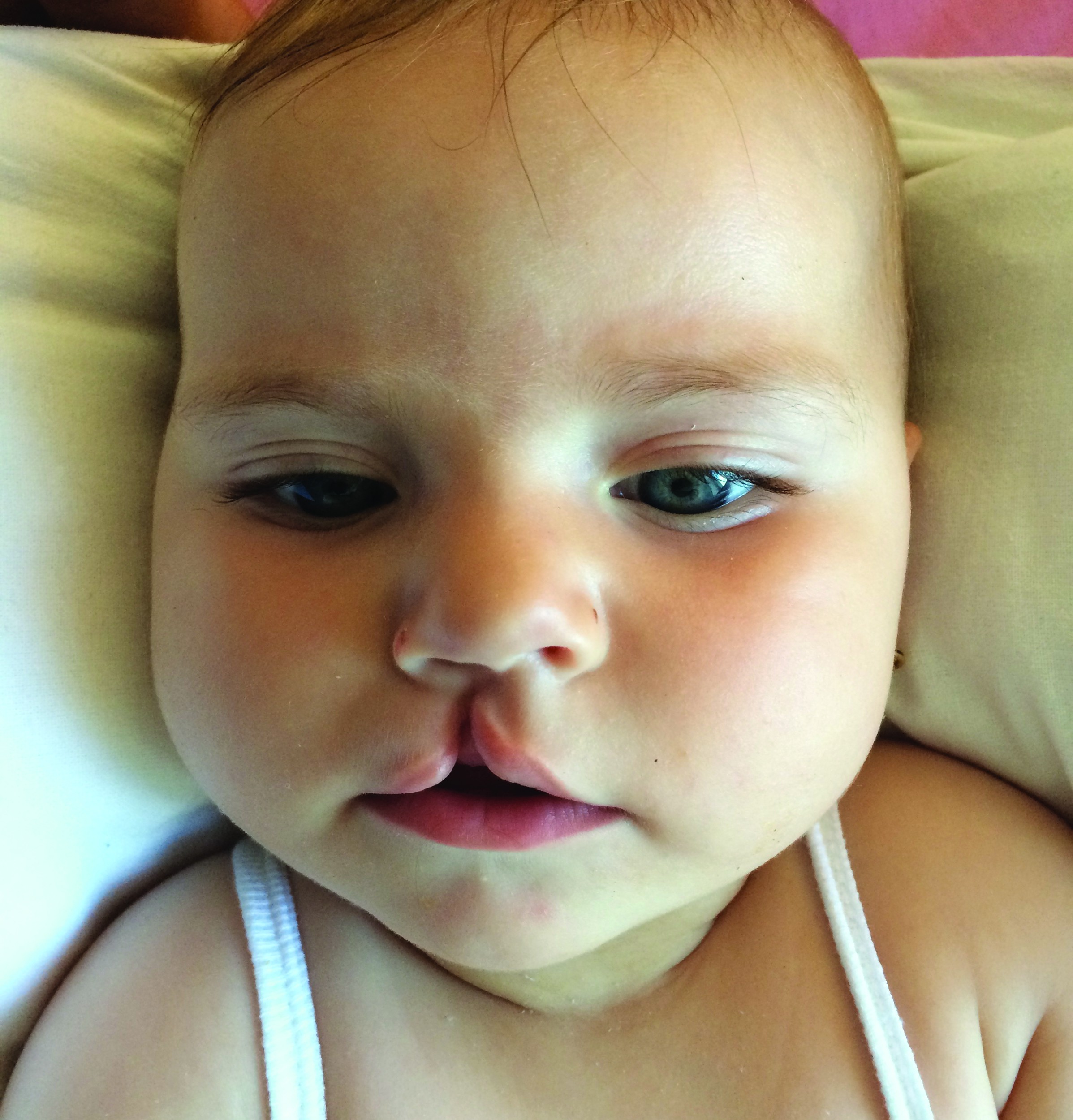
Diviziunea buzei superioare (cheilodisrafiile buzei, despicătura labială), cunoscută şi sub numele de „buză de iepure”, poate interesa buza parţial (doar o indentaţie la nivelul buzei) sau în întregime, inclusiv deschiderea narinei (din Error! Hyperlink reference not valid.: keilon - buză şi schizis - separare). Diviziunea labială este mai frecventă la sexul masculin, respectiv 60-70% din cazuri, un rol în apariţia sa avându-l şi vârsta înaintată a mamei sau anumiţi factori hormonali(5). Este datorată lipsei de sudură între mugurele maxilar şi nazal medial. Frecvent este unilaterală (figura 1), de obicei pe partea stângă(4), dar poate fi şi bilaterală, în special în cadrul unor sindroame sau al trisomiilor. În 80% din cazuri, despicătura labială este unilaterală şi doar în 20% din cazuri este bilaterală. Interesează partea cărnoasă a buzei superioare şi mai rar poate interesa şi partea osoasă a maxilei între incisiv şi canin, uneori ajungând până la canalul nazopalatin. Buza afectată prezintă toate elementele anatomice, permiţând reconstituirea, dar acestea sunt deplasate şi uneori nedezvoltate corespunzător, întotdeauna existând o hipoplazie regională. Diviziunea labio-maxilară (cheilo-gnato-disrafia, despicătura labio-maxilară) apare atunci când diviziunea labială se asociază cu diviziunea totală sau parţială a palatului anterior; poate fi uni- sau bilaterală.
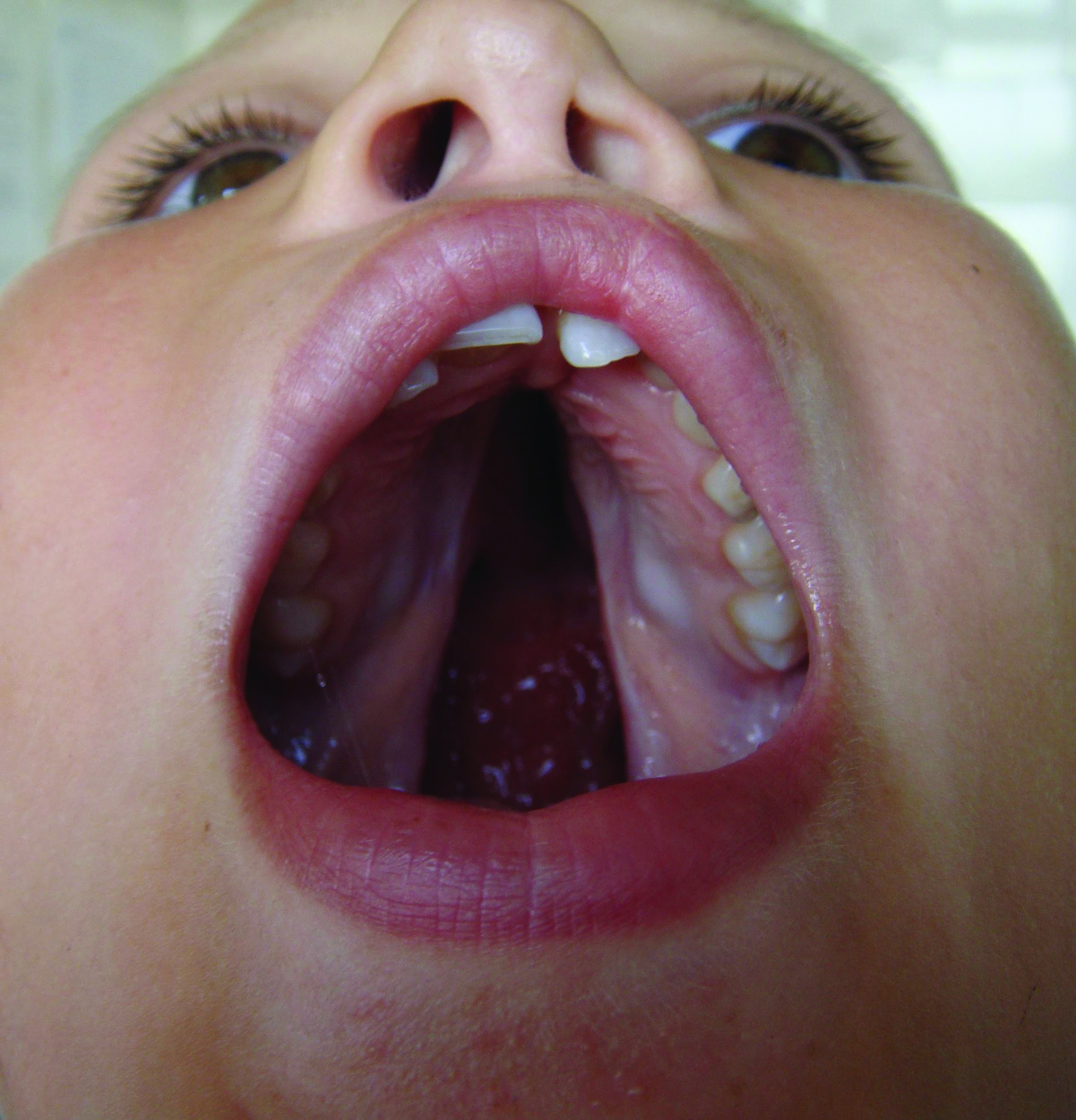
Diviziunile palatului posterior pot fi şi izolate, fără diviziune labială, pot interesa palatul dur în întregime sau parţial şi se numesc diviziuni velo-palatine (stafilo-urano-disrafii, despicături palatine). Dacă interesează numai palatul moale (lueta şi vălul palatin), atunci acest tip de diviziuni se numesc diviziuni velare (stafilo-disrafii, lueta bifida). În diviziunile velo-palatine, vomerul poate fi alipit unei lame palatine (diviziunea palatină unilaterală), iar în diviziunea palatină bilaterală rămâne liber, nealipit. Despicăturile palatine izolate sunt mai frecvente la sexul feminin, au o incidenţă de 4/10.000 de nou-născuţi şi sunt frecvent asociate cu alte malformaţii congenitale(3).
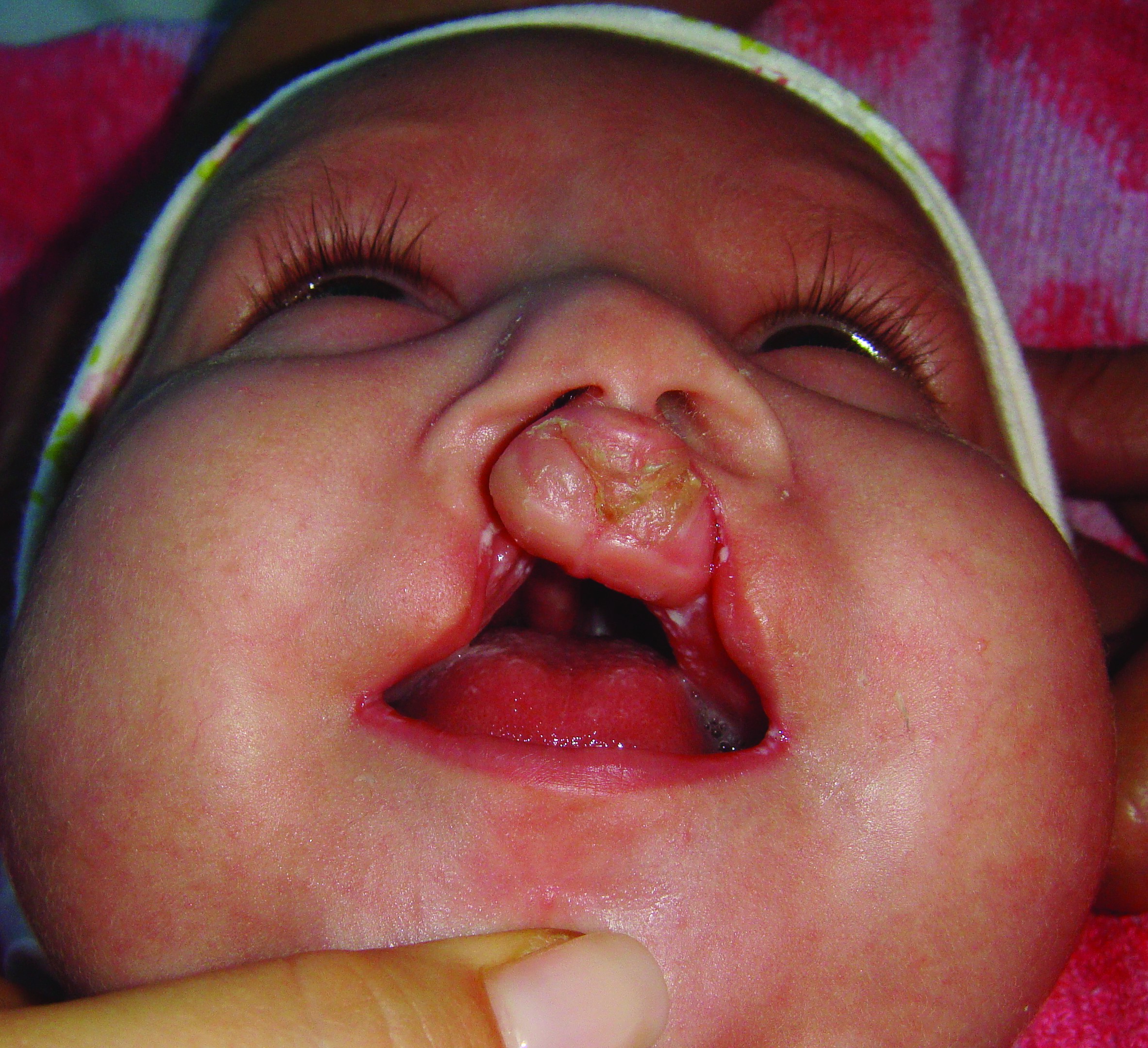
Când diviziunea interesează totalitatea palatului dur şi moale şi buza superioară, se numeşte diviziune labio-maxilo-palatină (cheilo-gnato-palato-disrafie); poate fi unilaterală sau bilaterală, varianta din urmă fiind cunoscută şi sub numele de „gură de lup” (figurile 2 și 3). În astfel de cazuri, cavitatea bucală poate comunica cu cavitatea nazală, împiedică suptul la sugar, lichidele refulează pe nas, existând riscul de aspiraţie, şi apar frecvente infecţii de căi respiratorii superioare. De asemenea, din cauza diastazisului osos, rapoartele anatomice sunt modificate, trompa lui Eustachio este mai scurtă şi orizontalizată, făcând facilă comunicarea microbiană între faringe şi urechea medie, de aici riscul mărit al infecţiilor otice. Fonaţia este, de asemenea, afectată, fiind recomandată protezarea palatului dur cu mulaje din silicon, iar tratamentul logopedic este recomandat încă din perioada de sugar.
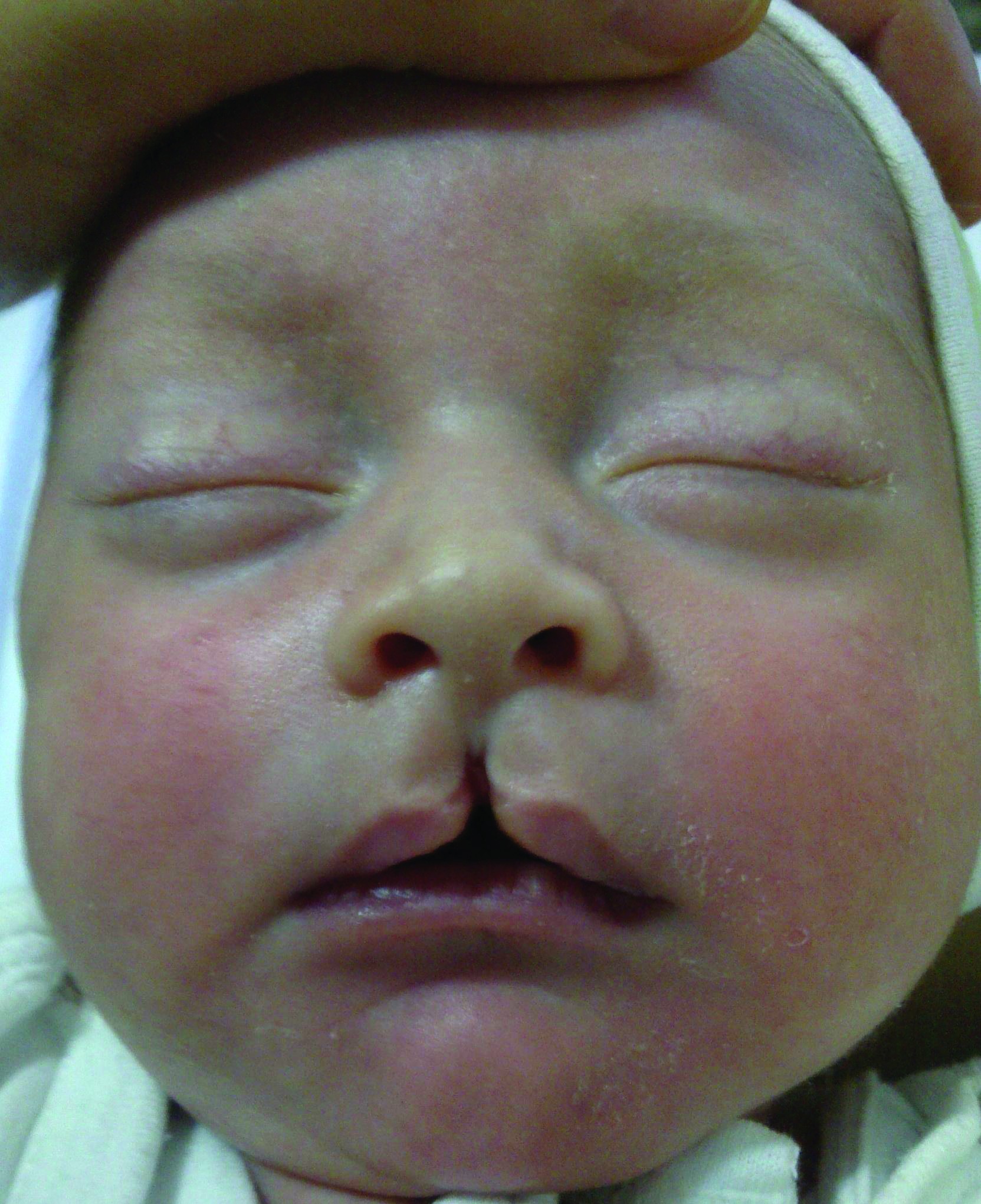
Diviziunea mediană a buzei superioare (schizocefalia) se datoreşte lipsei de fuziune a mugurilor nazali mediali şi lipsei formării osului intermaxilar. Această malformaţie este foarte rară şi reprezintă un simptom patognomonic al sindromului Mohr, sindrom ce se transmite autozomal recesiv. Această anomalie reprezintă, de fapt, adevărata „buză de iepure” (figura 4). Diviziunea mediană a buzei inferioare este foarte rară şi se datorează lipsei masei mezenchimale dintre mugurii mandibulari.
Despicăturile labio-maxilo-palatine pot fi izolate, dar în aproximativ 37% din cazuri sunt asociate cu alte anomalii congenitale, cele mai frecvente fiind cardiace, iar uneori fac parte din cadrul unor sindroame malformative (sindrom van der Woude, Pierre-Robin, Opitz, Treacher-Collins, Smith-Lemli-Opitz), ceea ce demonstrează componenta genetică a acestor afecţiuni, sau pot fi expresia unor trisomii, precum trisomia 18(6,7). Asocierea malformaţiilor congenitale şi a anomaliilor cromozomiale este responsabilă de mortalitatea de 10-15% asociată despicăturilor labiale şi palatine(8).
Diagnosticul se poate stabili încă din perioada antenatală, din primele 13-16 săptămâni de gestaţie, cu ajutorul ecografiei uterului gravid sau al RMN-ului fetal, importanţa diagnosticului antenatal constând atât în pregătirea psihologică a cuplului parental, în trimiterea pacientului imediat postnatal către un centru multidisciplinar, specializat în tratamentul unor astfel de anomalii, cât şi mai ales în diagnosticarea eventualelor anomalii asociate grave, uneori fiind chiar indicat avortul terapeutic (ca în cazul trisomiilor 13 sau 18). Cele mai multe despicături sunt însă diagnosticate la naştere, la simpla inspecţie a nou-născutului; în caz de despicătură palatină izolată, la tentativa de alimentaţie, se observă refluarea lichidelor pe nas sau dificultăţi ale alimentaţiei la sân din cauza imposibilităţii asigurării etanşeităţii orale şi a mecanismului de pompă. În sindromul Pierre-Robin, din cauza hipoplaziei mandibulare asociate şi a protruziei posterioare a limbii, pot apărea şi dificultăţi de respiraţie. Cu timpul, aceşti copii pot dezvolta frecvent infecţii respiratorii repetate, adenoidite, otite medii şi chiar pierderea auzului, având nevoie de consulturi şi intervenţii în sfera ORL, vor prezenta tulburări de fonaţie, necesitând terapie logopedică, iar anomaliile dentare vor necesita protezare, intervenţii stomatologice şi OMF.
Malformaţia este aşadar invalidantă prin consecinţele ei pe multiple planuri: estetic, psihologic, al vorbirii, al nutriţiei, al malformaţiilor asociate, de aceea managementul terapeutic al acestei afecţiuni începe cu diagnosticul antenatal şi continuă imediat postnatal, într-o echipă multidisciplinară alcătuită din genetician, pediatru, chirurg pediatru, chirurg plastician, medic oro-maxilo-facial, stomatolog, ortodontist, logoped, medic otorinolaringolog, psihoterapeut(2). Numai prin colaborarea tuturor acestor specialişti se va reuşi corecţia aspectului estetic al copilului, lucru foarte important pentru familie şi, nu în ultimul rând, se vor asigura prevenirea infecţiilor otice, prevenirea infecţiilor respiratorii, asigurarea premiselor pentru o vorbire normală, pentru corecţia dentiţiei, ceea ce va duce implicit şi la o alimentaţie normală. Tratamentul modern al diviziunilor labio-maxilo-palatine este complex şi trebuie realizat în centre specializate; trebuie început de la naştere şi poate dura până la adolescenţă sau până la vârsta adultă, implicând prezenţa unei întregi echipe de specialişti, care îşi va aduce contribuția la tratamentul diverselor aspecte patologice ale acestor malformaţii(9,10).
În ceea ce priveşte principiile tratamentului chirurgical, acestea implică reconstituirea şanţului narinar şi a planşeului foselor nazale, reconstrucţia arcului maxilar dacă acesta este afectat, repoziţionarea aripii nazale, reconstrucţia buzei (buza albă, mucoasa roşie, musculatura - orbicularul buzei, mucoasa vestibulară), repararea breşei palatine şi refacerea anatomiei velare cât mai aproape de normal. Cronologia clasică implică efectuarea cheilo-plastiei ca intervenţie primară în jurul vârstei de 3-6 luni pentru formele unilaterale şi 6-8 luni pentru formele bilaterale, uranostafilorafia la vârsta de 18-24 de luni, urmând apoi rinoplastia, corecţia cicatricelor, închiderea diviziunii maxilare, eventual cu utilizarea de grefoane osoase recoltate din creasta iliacă, alte corecţii care ţin de chirurgia ortognatică după vârsta de 12 ani(11). În cazul abordării tratamentului în secvenţă inversată, în jurul vârstei de 3 luni se practică veloplastia precoce, care permite o creştere normală în ansamblu a structurilor afectate, iar la vârsta de 6 luni se practică în acelaşi timp operator repararea buzei şi a palatului osos, repararea acestuia din urmă fiind punctul cel mai delicat al intervenţiei. Este încă în vigoare în unele centre „regula lui 10”, iniţiată de către Wilhelmsen încă din 1966, respectiv iniţierea tratamentului chirurgical atunci când copilul împlineşte vârsta de 10 săptămâni, are hemoglobina peste 10 g/dl, leucocitele sunt sub 10.000/mm3, iar greutatea este peste 10 lbs (4,5 kg)(12).
Tratamentul ortodontic, dacă este necesar, trebuie început de la vârsta de 3 luni, iar terapia logopedică, de la vârsta de 9 luni. Toate intervenţiile medicale şi chirurgicale au drept scop redarea unui aspect estetic şi morfologic cât mai apropiat de cel normal, precum şi asigurarea dezvoltării psihosomatice şi emoţionale normale(13). Evoluţia pe termen lung este favorabilă în cazul preluării pacientului încă din perioada prenatală sau imediat postnatală de o echipă multidisciplinară care îl va urmări până la vârsta adultă, asigurându-i accesul la toate stadiile tratamentului medical, chirurgical, precum şi consilierea psihologică necesară integrării în societate şi asigurării respectului de sine.
Bibliografie
2. Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009; 374(9703):1773–8510. 1016/S0140-6736(09)60695-4.
3. Mahdi A. Shkoukani, Michael Chen, Angela Vong. Cleft Lip – A Comprehensive Review. Front Pediatr. 2013; 1:53. Prepublished online 2013 Nov 13. Published online 2013 Dec 27. doi: 10.3389/fped.2013.00053.
4. Yoshikazu Nagase, Nagato Natsume, Tomoki Kato, Toko Hayakawa. Epidemiological Analysis of Cleft Lip and/or Palate by Cleft Pattern. J Maxillofac Oral Surg. 2010 Dec; 9(4): 389–395. Published online 2011 Mar 11. doi: 10.1007/s12663-010-0132-6.
5. James WH. Are oral clefts a consequence of maternal hormone imbalance? Evidence from the sex ratios of sibs of probands. Teratology. 2000; 62(5):342–510. 1002/1096-9926(200011)62:5<342::AID-TERA8>3.0.CO;2-8.
6. Setó-Salvia N, Stanier P. Genetics of cleft lip and/or cleft palate: association with other common anomalies. Eur J Med Genet. 2014 Aug; 57(8):381-93. doi: 10.1016/j.ejmg.2014.04.003. Epub 2014 Apr 21.
7. Calzolari E, Pierini A, Astolfi G, Bianchi F, Neville AJ, Rivieri F. Associated anomalies in multi-malformed infants with cleft lip and palate: an epidemiological study of nearly 6 million births in 23 EUROCAT registries. Am J Med Genet. 2007; 143:528–3710.1002/ajmg.a.31447.
8. Fogh-Anderson P. Incidence of cleft lip and palate: constant or increasing? Acta Chir Scand. 1961; 122:106-11.
9. Stock NM, Feragen KB, Rumsey N. “It Doesn’t All Just Stop at 18”: Psychological Adjustment and Support Needs of Adults Born With Cleft Lip and/or Palate. Cleft Palate Craniofac J. 2014 Nov 5. [Epub ahead of print].
10. Hartzell LD, Kilpatrick LA. Diagnosis and management of patients with clefts: a comprehensive and interdisciplinary approach. Otolaryngol Clin North Am. 2014 Oct; 47(5):821-52. doi: 10.1016/j.otc.2014.06.010.
11. Farronato G, Kairyte L, Giannini L, Galbiati G, Maspero C. How various surgical protocols of the unilateral cleft lip and palate influence the facial growth and possible orthodontic problems? Which is the best timing of lip, palate and alveolus repair? Literature review. Stomatologija. 2014; 16(2):53-60.
12. Wilhelmsen HR, Musgrave RH. Complications of cleft lip surgery. Cleft Palate J. 1966; 3:223-31.
13. Talmant JC, Talmant JC. Cleft rhinoplasty, from primary to secondary surgery. Ann Chir Plast Esthet. 2014 Sep 23. pii: S0294-1260(14)00131-9. doi: 10.1016/j.anplas.2014.08.004. [Epub ahead of print].
Articole din ediţiile anterioare
Hemangiomul infantil − posibilităţi de abordare terapeutică
tumori vasculare, malformaţii vasculare, hemangiom, propranolol, scleroterapie
Managementul şocului septic la copil: controverse şi certitudini
În ciuda progreselor înregistrate în vaccinologie, antibioterapie şi terapie intensivă, sepsisul şi şocul septic la copil rămân o cauză majoră...
Aspecte clinice şi hematologice la pacienţii pediatrici diagnosticaţi cu anemie Fanconi – experienţa unui centru
Sindroamele de insuficienţă medulară congenitală reprezintă un grup de afecţiuni hematologice rare care se caracterizează prin insuficienţă medular...
Studiu prospectiv privind managementul durerii acute la nou-născutul cu afecţiuni chirurgicale prin corelaţia scalei de durere CRIES cu protocolul terapeutic etapizat
The last 2-3 decades have brought major changes in the management of pain in the newborn.