Polymyositis is a rare disease from the idiopathic inflammatory myopathies group, characterized by the appearance in the skeletal muscle of an inflammatory process that causes progressive weakness in the proximal muscles of the limbs. We report the case of a 13-year-old female known with polymyositis, whose evolution is characterized by several outbursts of the disease despite the administration of immunosuppressive therapy.
PAGINA REZIDENTULUI
Polimiozita - caz clinic
Polymyositis - a clinical case
First published: 04 aprilie 2017
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Pedi.45.1.2017.593
Abstract
Rezumat
Polimiozita este o boală rară din grupul miopatiilor inflamatorii idiopatice, caracterizată prin apariţia la nivelul muşchiului scheletic a unui proces inflamator ce determină ulterior slăbiciunea progresivă a musculaturii proximale a membrelor. Prezentăm cazul unei paciente de 13 ani cunoscută cu polimiozită, care se remarcă printr-o evoluţie marcată de numeroase puseuri ale bolii, în ciuda tratamentului imunosupresiv administrat.
Prezentarea cazului
Pacienta M.A., în vârstă de 13 ani, din mediul rural, judeţul Vrancea, aflată în evidenţa Clinicii II Pediatrie a Spitalului Clinic de Urgenţă pentru Copii „Sf. Maria” din Iaşi cu polimiozită, insuficienţă mitrală grad I-II şi insuficienţă tricuspidiană grad I-II, se internează pentru reevaluare clinico-biologică pe data de 30.01.2017.
Antecedentele heredo-familiale ale pacientei sunt nesemnificative - nu există boli cronice sau cu transmitere congenitală, fără contact TBC.
Istoricul bolii actuale debutează în 2013, când pacienta se prezintă din proprie iniţiativă la spitalul din Focşani pentru scădere semnificativă a forţei musculare, MS, MI, mialgii distale MI, fenomen Raynaud la nivelul mâinilor şi picioarelor la expunerea la frig. După 3 zile de spitalizare, pacienta este transferată la Spitalul Clinic de Urgenţă pentru Copii „Sf. Maria” din Iaşi, unde în urma investigaţiilor clinice şi paraclinice se stabileşte diagnosticul de polimiozită, insuficienţă mitrală grad I-II şi insuficienţă tricuspidiană grad I-II. În 2013 este iniţiată terapia cu prednison 10 mg/zi per os, discontinuu, iar în 2014 schema terapeutică este modificată - prednison 10 mg/zi alternativ cu Imuran 50 mg/zi per os, menţinută până în prezent. Evoluţia bolii în această perioadă a fost marcată de numeroase perioade de remisiuni şi recăderi pentru care pacienta a avut un număr mare de internări, pe durata cărora a primit pulsaţii cu metilprednisolon i.v.
Examenul clinic efectuat la internare relevă următoarele elemente patologice: eritem facial şi la nivelul feţei dorsale a mâinilor, eritem al extremităţii degetelor MI şi semnul Gottron prezent, suflu sistolic în focarul mitralei şi tricuspidei, apetit diminuat, cavitate bucală - carii dentare.
În cadrul investigaţiilor paraclinice şi biologice, am evaluat sindromul inflamator (VSH=24 mm/h, CRP semicant >6 mg/dl, Fibrinogen=253 mg/dl), sindromul hematologic (Hb=10,3 g/dl, Tr=481x10³/mm³, D-dimeri=1749 microg/L), sindromul muscular (CPK=660 U/l, LDH=643 U/l, TGO=139 U/l↑; Normal: 5-37 U/l; creatină urinară=10,4 mg/kg/24h) şi sidromul imunologic (FR=256 ui/l, celule lupice - absente, Ac anti-nucleari - negativi, Ac anti-ADNdS Ig G ELISA =5,7403 UI/ml, crioglobuline - absente, Ig A, Ig G, Ig M în parametri normali, Ac anti-U1RNP, Ac anti-SCL70, Ac anti-centromer, Ac anti-Jo1 nu au fost efectuaţi).
Apectul electrocardiogramei este normal, iar consultul ecocardiografic susţine diagnosticul de insuficienţă mitrală grad I-II şi insuficienţă tricuspidiană grad I-II, cu care este cunoscută pacienta.
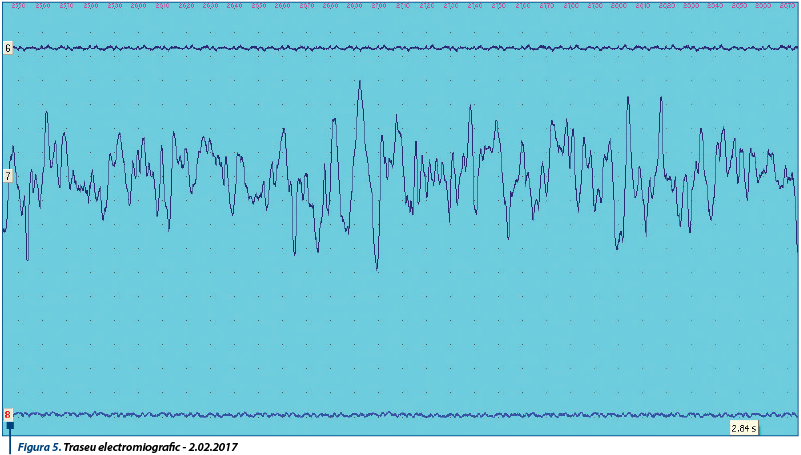
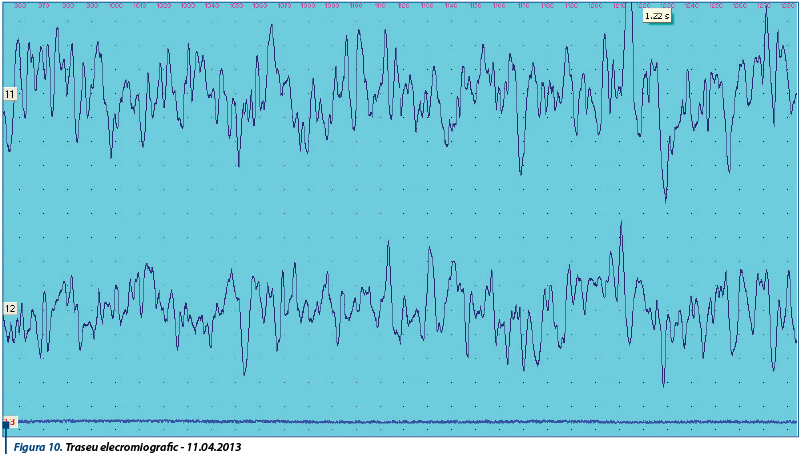
Electromiograma prezintă un traseu specific de miopatie, cu multiple polivârfuri diseminate nesistematizate şi potenţiale de unitate motorie de diverse morfologii şi amplitudini.
Criterii de diagnostic al polimiozitei:
1. Oboseală simetrică a musculaturii proximale.
2. Creşterea valorii enzimelor musculare serice: CK, aldolază, LDH, AST, ALT.
3.Modificări electromiografice de tip miopatic: unde largi, fibrilaţii spontane, unităţi motorii polifazice de scurtă durată, amplitudine joasă, descărcări repetitive cu frecvenţă înaltă.
4. Biopsie musculară caracteristică: degenerare/regenerare a miofibrelor, infiltrat interstiţial cu mononucleare, atrofie perifasciculară. Diferenţa între PM şi DM o realizează tipul infiltratului şi localizarea lui, în interiorul fibrei sau perifascicular, în peretele vascular.
Deşi pacienta a fost diagnosticată cu polimiozită la prima prezentare în clinică, prin îndeplinirea a 3 criterii din 4, la internarea actuală prezintă la examenul clinic semnul Gottron, specific dermatomiozitei. Acest fapt ne pune în dificultate pentru a stabili un diagnostic cu certitudine, iar investigaţia utilă în determinarea diagnosticului (biopsia musculară) a fost refuzată de către aparţinătorii pacientei.
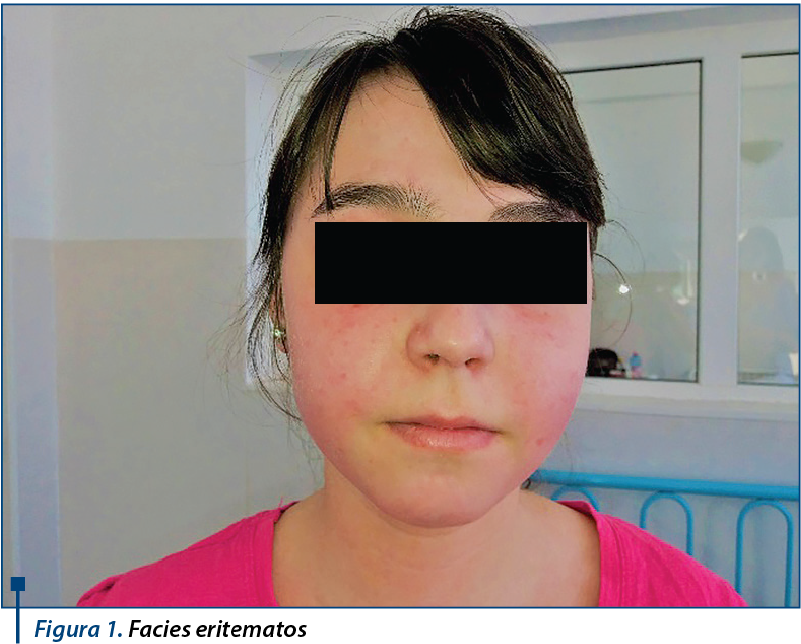
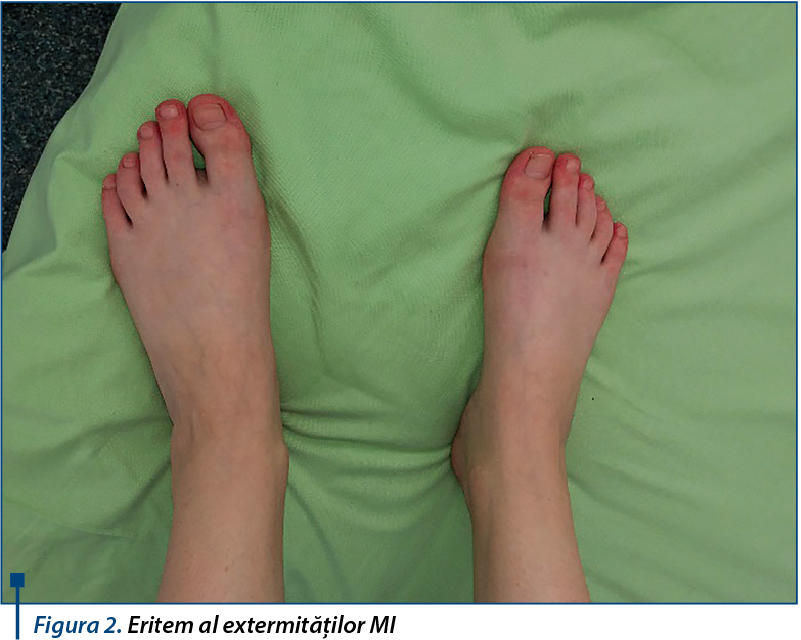
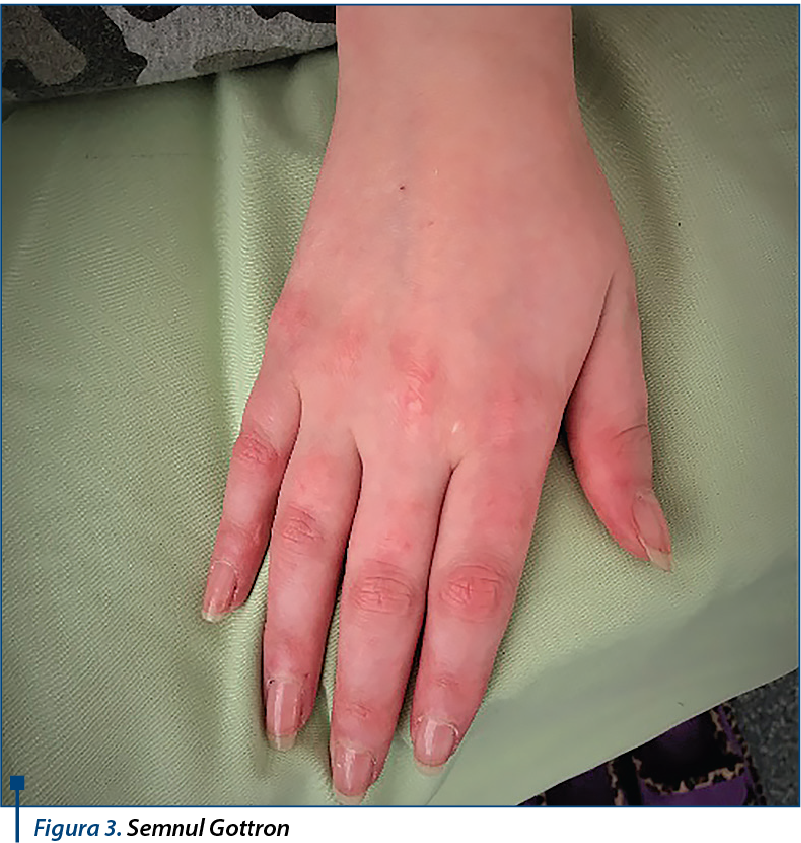

Aşadar, coroborând datele prezentate, se susţine diagnosticul pozitiv de:
1. Polimiozită în plin puseu.
2. Spasmofilie hipocalcemică.
3. Anemie hipocromă microcitară.
4. Insuficienţă mitrală grad I-II.
5. Insuficienţă tricuspidiană grad I-II.
Tratament
La tratamentul de fond imunosupresiv urmat de pacientă (prednison 10 mg/zi alternativ cu Imuran-Azatioprină 50 mg/zi per os), din cauza puseului bolii, pe perioada internării s-a iniţiat pulsaţia de metilprednisolon i.v. 1 g/zi x 3 zile.
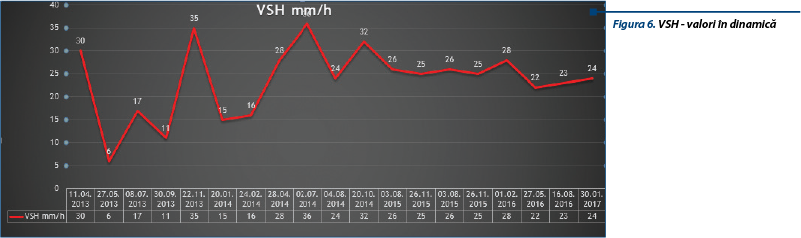
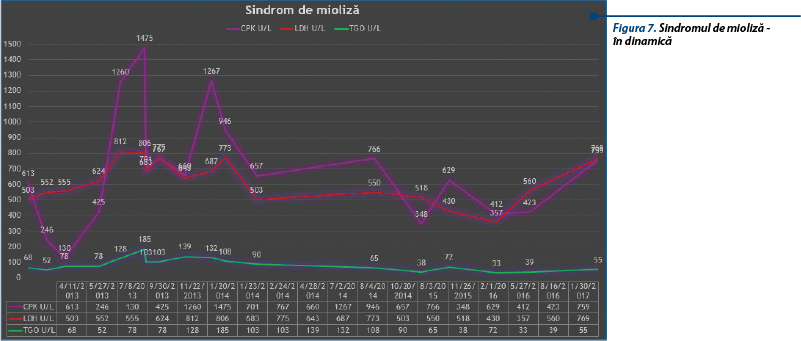
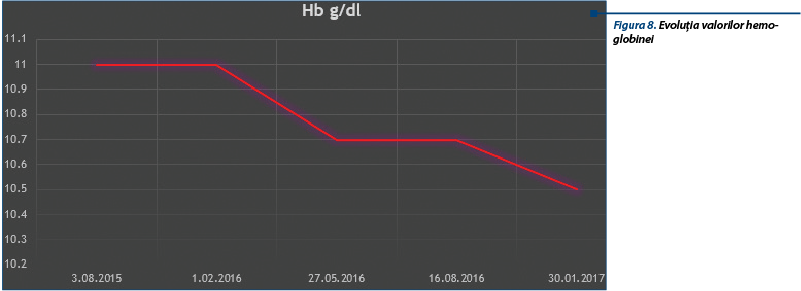
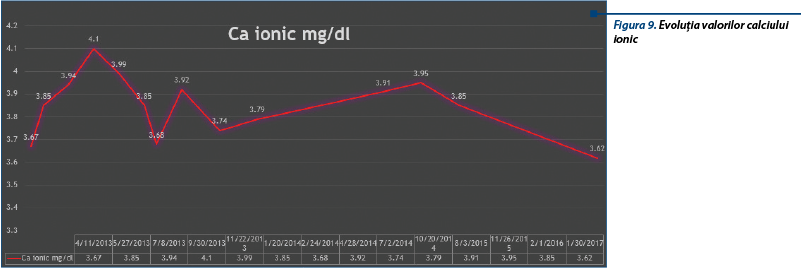
Evoluţia bolii
În urma analizei investigaţiilor efectuate, prezentăm evoluţia în dinamică a următorilor parametri biologici:
Numeroasele puseuri ale bolii s-au însoţit de creşteri ale parametrilor ce definesc sindromul inflamator, precum şi de creşteri marcante ale nivelului seric al enzimelor musculare. Se observă remiterea fiecărui episod după administrarea endovenoasă a câte 3 pulsaţii consecutive de SoluMedrol (metilprednisolon).
Întreaga evoluţie a bolii a fost însoţită de prezenţa constantă a unei anemii hipocrome normocitare şi de o spasmofilie hipocalcemică, acestea nefiind corectate în urma tratamentului specific primit.
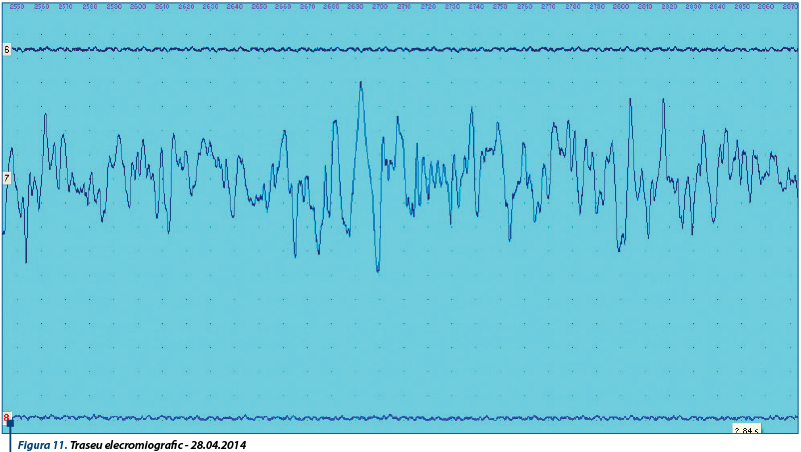
Explorarea electromiografică în timp arată aproximativ acelaşi aspect de traseu miogen, cu multiple polivârfuri diseminate, nesistematizate şi cu potenţiale de unitate motorie asimetrice, neregulate, de amplitudini şi durate diferite. Aspectul electromiografic, coroborat cu datele clinice şi paraclinice, susţine diagnosticul de miopatie inflamatorie cronică.
Prognosticul bolii
Iniţierea tratamentului la mai puţin de 6 luni de la diagnostic, răspunsul prompt la corticoterapie, vârsta pacientei mai mică de 50 de ani, lipsa afectării pulmonare şi a neoplaziilor sugerează un prognostic favorabil al bolii. Trebuie ţinut cont însă de frecventele recăderi apărute sub terapia imunosupresivă, ce recomandă acordarea unei atenţii sporite şi reevaluarea periodică a pacientei. Pe termen lung, trebuie urmărită eventuala apariţie a complicaţiilor legate de boală: agravarea oboselii musculare şi apariţia simptomatologiei extramusculare (boală pulmonară interstiţială, boală cardiacă, disfagie, malabsorbţie, neoplasm - frecvent pulmonar şi de sân), dar şi dezvoltarea unor efecte secundare ale terapiei cu prednison şi azatioprină.
Particularitatea cazului constă în evoluţia marcată de puseuri ale bolii, apărute în ciuda tratamentului supresiv corect administrat.
Bibliografie
1. Harris-Love MO, Shrader JA, Koziol D, Pahlajani N, Jain M, Smith M, et al. Distribution and severity of weakness among patients with polymyositis, dermatomyositis and juvenile dermatomyositis. Rheumatology (Oxford). 2009; 48:134–9.
2. Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013; 92:25–41.
3. Benveniste O, Romero NB. Myositis or dystrophy? Traps and pitfalls. Presse Med. 2011; 40:e249–55.
4. Khadilkar SV, Patil SG, Amin SN. Study of idiopathic inflammatory myopathies with special reference to borderland between idiopathic inflammatory myopathies and muscular dystrophies. Neurol India. 2008; 56:356–62.
5. Choy EH, Hoogendijk JE, Lecky B, Winer JB. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev 2005.
6. Airio A, Kautiainen H, Hakala M: Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol 2006, 25:234-239.
2. Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013; 92:25–41.
3. Benveniste O, Romero NB. Myositis or dystrophy? Traps and pitfalls. Presse Med. 2011; 40:e249–55.
4. Khadilkar SV, Patil SG, Amin SN. Study of idiopathic inflammatory myopathies with special reference to borderland between idiopathic inflammatory myopathies and muscular dystrophies. Neurol India. 2008; 56:356–62.
5. Choy EH, Hoogendijk JE, Lecky B, Winer JB. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev 2005.
6. Airio A, Kautiainen H, Hakala M: Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol 2006, 25:234-239.