Osteochondrodysplasias (OCD) are a large heterogenous group of bone diseases. Also known as skeletal dysplasias, they can have genetic causes or they can be the result of exogenous factors. The last classification of OCD includes 375 diseases. Ultrasonographic findings, computed tomography scan and magnetic resonance imaging scan can help prenatal diagnostic. Genetic testing is the most important diagnostic tool. In Romania there are two laws regarding abortion. The abortion decision belongs to the parents as long as their decision respects the law. Despite the physical aspect, these patiens can have a normal inteligence. Efforts must be done for the social integration of these patients.
Aspecte medico-legale în osteocondrodisplaziile neletale
Medico-legal aspects in non-lethal osteochondrodysplasia
Elena Alina Bordea,
Diana-Elena Comandașu,
C. Coroleucă,
Octavia Velicu,
Prof. Dr. Elvira Brătilă,
Mihai Mitran
First published: 30 octombrie 2017
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.1.3.2017.1176
Abstract
Rezumat
Osteocondrodisplaziile (OCD) sunt un grup larg, heterogen, de anomalii care implică formarea şi creşterea oaselor. Numite şi displazii scheletale, se pot manifesta încă din stadiile precoce ale dezvoltării fetale şi pot fi cauzate de factori exogeni sau pot avea determinism genetic. Clasificarea OCD include aproximativ 375 de boli grupate în peste 30 de grupuri majore. Diagnosticul prenatal este în principal ecografic, iar RMN-ul, CT-ul şi testarea genetică sunt folositoare pentru susţinerea diagnosticului prezumtiv. Legile care fac referire la întreruperea sarcinii se găsesc în Codul penal, articolele 201-202. Pacienţii cu displazii scheletale neletale pot avea un intelect normal, în ciuda aspectului fizic. Decizia de întrerupere a sarcinii la un făt cu OCD le aparţine în totalitate părinţilor atât timp cât respectă cadrul legal. Trebuie făcute eforturi pentru acceptarea şi integrarea acestor pacienţi în societate.
Introducere
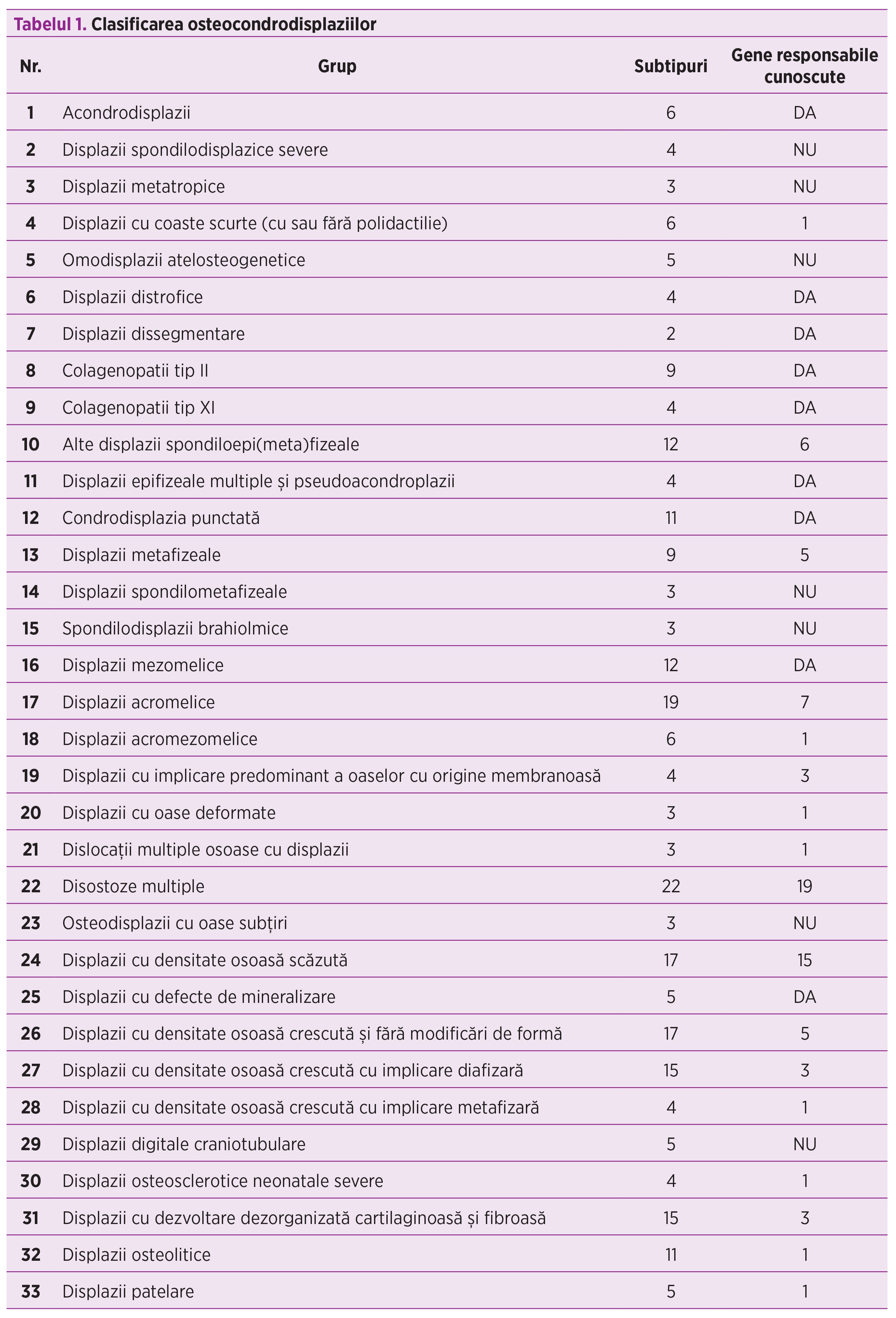
Osteocondrodisplaziile constituie un grup larg, eterogen, de anomalii care implică formarea şi creşterea oaselor. O parte dintre ele sunt asociate cu anomalii de organ şi sunt letale din cauza insuficienţei respiratorii. Osteocondrodisplaziile, numite şi displazii scheletale, se pot manifesta încă din stadiile precoce ale dezvoltării fetale şi pot fi cauzate de factori exogeni, precum expunerea la substanţe teratogene, sau pot fi determinate de cauze intrinseci, precum cauzele genetice(1).Cea mai recentă clasificare a osteocondrodisplaziilor include aproximativ 375 de boli grupate în peste 30 de grupuri majore (tabelul 1) în funcţie de criterii moleculare, biochimice şi imagistice(2,17). Un număr de 215 dintre displaziile scheletale sunt asociate cu mutaţii în 145 de gene(3).
Prevalenţa displaziilor scheletale este estimată la 2,4 la 10000 de naşteri. 5% dintre bolile genetice identificate la nou-născut sunt reprezentate de displaziile scheletale. Prevalenţa în sarcinile supravegheate cu morfologii fetale este de 7,5/10000(4).
Cele mai frecvente tipuri de osteocondrodisplazii sunt: displazia tanatoforică, acondroplazia, osteogeneza imperfectă şi acondrogeneza(4). Displazia tanatoforică şi acondrogeneza sunt responsabile de 62% dintre toate displaziile scheletale letale. Cea mai frecventă formă de osteocondrodisplazie neletală este acondroplazia(5).
Acondroplazia prezintă o incidenţă între 5 şi 15 la 100000 de naşteri şi este cauzată de o mutaţie a genei FGFR3 localizate pe cromozomul 4p16.3. Majoritatea cazurilor sunt sporadice şi rezultă de novo dintr-o mutaţie genică paternă. Diagnosticul este evident după 24 de săptămâni de gestaţie, atunci când ecografic se poate decela scurtarea oaselor lungi.
Acondroplazia este caracterizată de membre scurte cu degete scurte, iar la nivel facial se descrie o frunte largă şi proeminentă (bosa frontală) cu bază nazală aplatizată. Speranţa de viaţă şi performanţa intelectuală sunt bune. Diagnosticul poate fi confirmat prenatal prin analiza ADN-ului fetal din sângele matern, în acest caz căutându-se mutaţii ale genei FGFR3. Poate fi necesar tratament ortopedic, în scop corectiv(6).
Osteogeneza imperfectă este caracterizată de hipomielinizarea oaselor şi sclera albastră, iar tipul IV este cea mai uşoară formă dintre aceste boli de colagen ereditare. La pacienţii cu osteogeneză imperfectă tipul IV, fracturile nu sunt frecvente, iar sclera devine albă, în timp. Nu există asimetrie sau diformitate a membrelor, deşi pacienţii care suferă de această boală pot avea statură redusă. Boala este transmisă autozomal dominant şi nu poate fi diagnosticată de obicei decât după naştere(7).
Displazia spondilo-epifizară congenitală este asociată cu membre scurte, coloană vertebrală scurtă şi statură de aproximativ 120 cm.
Diagnosticul se face prin ecografie antenatală, iar boala este compatibilă cu o supravieţuire lungă, deşi apar slăbiciune musculară, restricţie în mobilizarea articulaţiilor şi deformarea progresivă a coloanei vertebrale, care determină cifoscolioza(6).
Reducerea izolată a membrelor este cel mai frecvent de cauză nongenetică, însă în unele cazuri poate face parte din anumite sindroame genetice. Este necesară efectuarea cariotipului fetal atunci când o reducere de dimensiune a unui membru este asociată cu alte anomalii nonscheletale. Prognosticul în caz de afectare unilaterală a unui membru este în general bun, în absenţa altor anomalii viscerale sau cromozomiale(8).
Dintre sindroamele specifice asociate cu reducerea izolată a unui membru fac parte: sindromul Brachmann de Lange, trombocitopenia asociată cu absenţa radiusului, trisomiile 13 şi 18, sindromul Holt-Oram, sindromul Fanconi, sindromul Robert’s, sindromul VATER, sindromul femur-fibulă-ulnă(6).
Metodologie - probleme de diagnostic
Diagnosticul prenatal este în principal ecografic, iar RMN-ul, CT-ul şi testarea genetică sunt folositoare pentru susţinerea diagnosticului prezumptiv(9).
Diagnosticul ecografic
Diagnosticul unei osteocondrodisplazii poate fi suspectat din cauza unei anomalii calitative osoase sau a unui femur scurt pentru vârsta gestaţională, la o examinare ecografică de rutină. În cazul unei suspiciuni diagnostice este necesară reevaluarea ecografică a întregului schelet fetal, pentru a determina care oase sunt afectate, tipul şi severitatea anomaliei(7).Scheletul fetal se dezvoltă relativ devreme în perioada fetală, aşadar diagnosticul prenatal al acestor boli este posibil. Osificarea are loc la vârste de gestaţie precoce: clavicula şi mandibula la 8 săptămâni, scapula la 12 săptămâni, iar metacarpienele şi metatarsienele la 12-16 săptămâni(10).
Scheletul fetal este vizibil la ecografia 2D care se realizează până la 12 săptămâni de gestaţie, acest lucru permiţând măsurarea humerusului şi a femurului fetale. Feţii care prezintă lungimea humerusului sau lungimea femurului sub percentila 5 până la 24 de săptămâni necesită evaluarea întregului schelet fetal şi sfat genetic. Multe displazii fetale prezintă disproporţii între măsurătorile craniului fetal, circumferinţa abdominală, mandibulă, claviculă, circumferinţa toracică şi lungimea oaselor lungi(11).
În afară de măsurarea ecografică a oaselor lungi există şi alţi parametri care ar trebui evaluaţi şi care pot fi utili în diferenţierea acestor boli. Printre aceşti parametri se numără profilul feţei, forma şi prezenţa corpurilor vertebrale şi aspectul relativ al mâinilor şi al picioarelor. Sunt multe tipuri de displazii scheletale asociate cu brahidactilie sau picior in varus equin(12).
Displaziile scheletale letale se instalează de obicei mai devreme şi au caracteristici fenotipice mai severe decât displaziile scheletale neletale, prin urmare displaziile scheletale letale sunt mai uşor de diagnosticat ecografic(13). Acurateţea diagnosticului este extrem de importantă, deoarece va afecta în mod semnificativ decizia de continuare a sarcinii, precum şi opţiunile de diagnostic prenatal la o sarcină viitoare. În studii retrospective, ecografişti cu experienţă au pus un diagnostic ecografic prenatal corect al displaziilor scheletale letale în 81% dintre cazuri şi în 31-78% dintre cazurile totale de osteocondrodisplazii. Aşadar, displaziile scheletale neletale sunt mai dificil de diagnosticat(14). Din acest motiv, osteocondrodisplaziile neletale sunt provocatoare din punct de vedere diagnostic.
Atunci când sunt suspectate anomalii la ecografia 2D, ultrasonografia 3D poate fi utilă în completarea diagnosticului. RMN-ul după 20 de săptămâni de gestaţie este o metodă complementară de diagnostic, care poate identifica anomalii ale coloanei vertebrale.
Probleme de diagnostic genetic
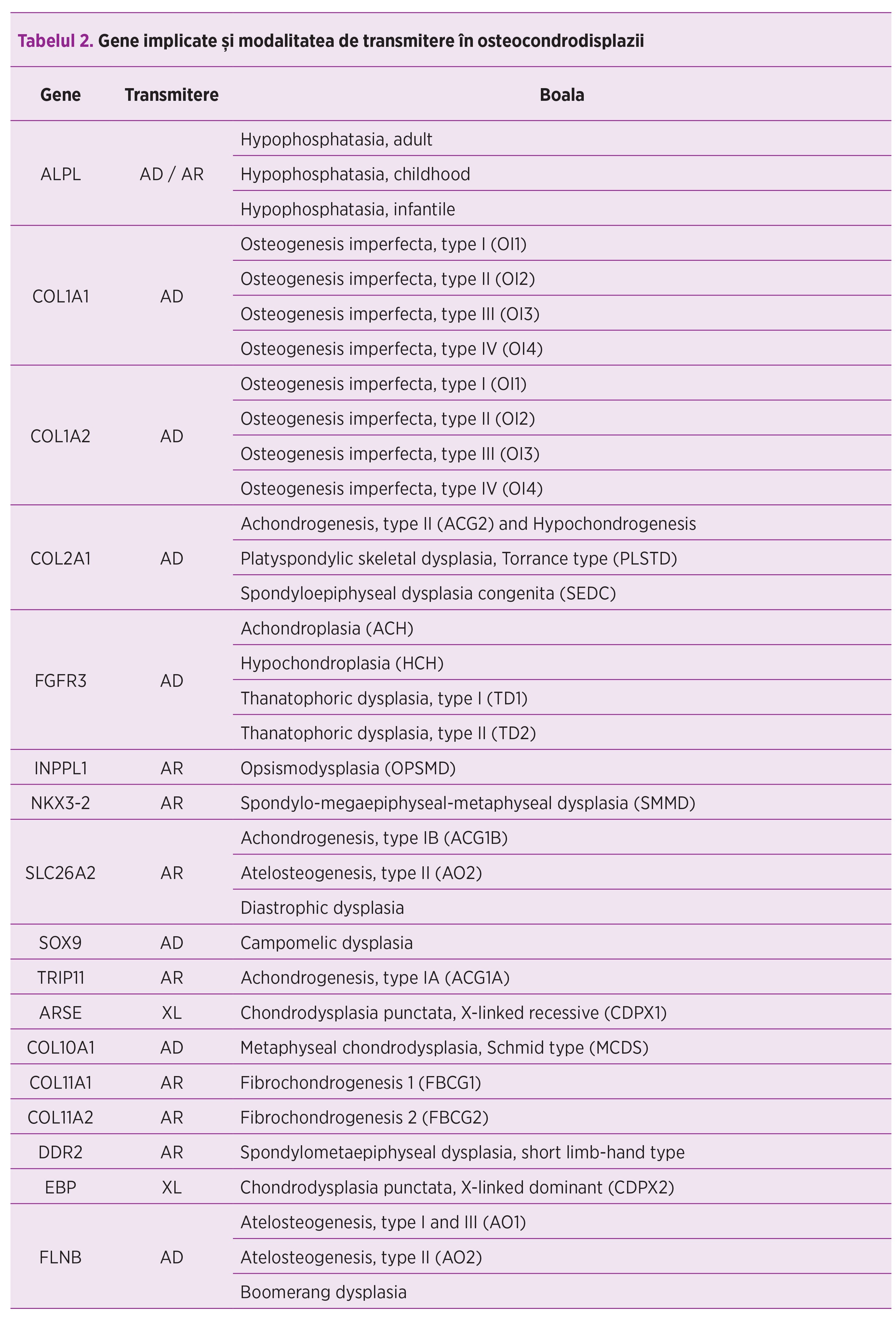
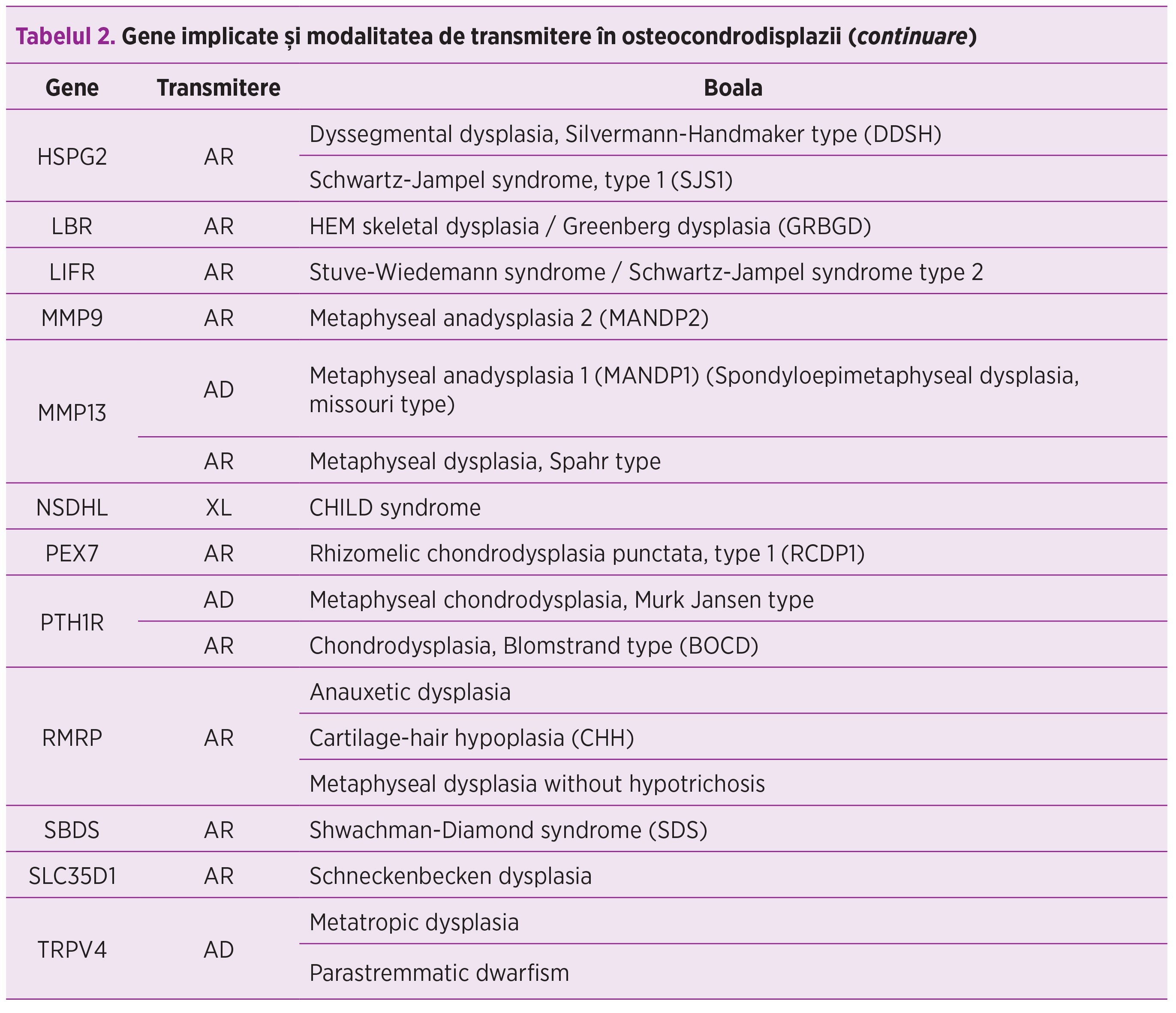
Ca în orice boală genetică, istoricul familial este important. Displaziile scheletale pot fi moştenite autozomal dominant, autozomal recesiv, pot fi X-linkate şi apar rar prin mecanisme care să includă deleţii, duplicaţii cromozomiale sau mozaicism(15).Procedurile pentru diagnosticul genetic sunt reprezentate de amniocenteză, biopsia de vilozităţi coriale şi determinarea ADN-ului fetal liber în sângele matern(16). Defecte genetice au fost identificate pentru aproximativ 50% dintre cele peste 350 de osteocondrodisplazii şi pot fi folosite în diagnosticarea precoce în cazul familiilor la risc(17).
Rolul testării moleculare la o sarcină normală cu risc sporadic de displazie scheletală este controversat. Testarea moleculară este justificată în caz de sarcini în care unul sau ambii părinţi sunt afectaţi de o boală scheletală cu transmitere autozomal dominantă. Acest lucru plasează fătul la risc pentru homozigoticitate sau heterozigoticitate, care sunt frecvent asociate cu mortalitate(16). Dacă ambii părinţi au acondroplazie, testarea pentru mutaţii ale genei FGFR3 este disponibilă. 97% dintre pacienţii cu acondroplazie suferă mutaţii identificabile. Dacă părinţii au displazii scheletale asociate cu mutaţii specifice, ar trebui încurajaţi să efectueze testare moleculară înaintea conceperii. Nu toate mutaţiile sunt identificabile(12).
Există paneluri de diagnostic genetic care sunt destinate detectării mutaţiilor la nivelul genelor responsabile de anumite displazii scheletice. Un număr de 29 de gene pot fi analizate. Acestea, împreună cu modalitatea de transmitere şi boala pe care o determină(19) sunt enumerate în tabelul 2.
Aspecte legislative - indicaţii de întrerupere a sarcinii
Legile care fac referire la întreruperea sarcinii se găsesc în Codul penal, articolele 201-202. Articolul 201, alineatul 6 menţionează: „Nu constituie infracţiune întreruperea cursului sarcinii în scop terapeutic efectuată de un medic de specialitate obstetrică-ginecologie, până la vârsta sarcinii de 24 de săptămâni, sau întreruperea ulterioară a cursului sarcinii, în scop terapeutic, în interesul mamei sau al fătului”. În plus, alineatul 7 al aceluiaşi articol menţionează faptul că: „Nu se pedepseşte femeia însărcinată care îşi întrerupe cursul sarcinii”.
Decizia de întrerupere a sarcinii la un făt cu osteocondrodisplazie neletală ridică mari dileme, iar decizia aparţine în totalitate părinţilor, atât timp cât respectă cadrul legal.
Consilierea cuplului
Atunci când ecografia şi metodele adiţionale de evaluare stabilesc diagnosticul de displazie scheletală neletală, acesta trebuie comunicat părinţilor. În multe cazuri, diagnosticul exact nu este cunoscut, însă prognosticul se bazează pe aspectul ecografic şi pe analiza genetică. Implicaţiile unei sarcini şi a naşterii unui copil cu această boală trebuie explicate părinţilor, pentru ca aceştia să poată lua o decizie informată referitoare la continuarea sarcinii. Opţiunile disponibile sunt de continuare a sarcinii, cu urmărirea cursului ei în echipă multidisciplinară (formată din medic obstetrician, medic neonatolog şi medic genetician), sau terminarea sarcinii, în cazul în care cadrul legal permite acest lucru.Din punct de vedere etic, decizia de întrerupere a sarcinii în cazul unui făt cu o displazie scheletală neletală, deci a unui făt malformat, dar cu un intelect normal, poate fi privită din două perspective.
Prima perspectivă susţine viaţa, adică indiferent de tipul de malformaţie, un produs de concepţie are dreptul la viaţă, indiferent dacă decizia ulterioară este de creştere a acestuia sau de oferire spre adopţie.
A doua perspectivă se referă la posibilitatea de alegere a părinţilor, fiind dreptul mamei să aleagă dacă îşi doreşte să nască un copil cu o malformaţie.
Osteocondrodisplaziile neletale sunt unice printre malformaţii, iar indivizii care supravieţuiesc pot avea o viaţă productivă, fără intervenţii chirurgicale sau tratament medicamentos. De exemplu, cu mii de ani în urmă, în Egipt, un nou-născut cu rinichi polichistic sau cu transpoziţie de mari vase ar fi murit, însă un copil cu acondrodisplazie ar fi supravieţuit şi ar fi stârnit interesul societăţii. Un bun exemplu în acest sens este pictura lui Velázquez „Las Meninas”, care ilustrează o imagine de la curtea regală a lui Filip al IV-lea, în care apare un copil ale cărui trăsături faciale şi membre scurte sunt tipice pentru acondrodisplazie.
Printre personalităţile care suferăsau au suferit de această patologie se numără:
- Henri de Toulouse-Lautrec - pictor francez din perioada postimpresionistă
- François de Cuvilliés - arhitect, cunoscut pentru Teatrul Cuvilliés din München
- Peter Dinklage - actor, cunoscut pentru rolul lui Tyrion Lannister din filmul Urzeala tronurilor
- Ellie Simmonds - înotătoare, a câştigat două medalii de aur la Jocurile Paralimpice de vară din 2008.
Concluzii
Osteocondrodisplaziile neletale, care au expresie fenotipică variabilă, sunt provocatoare din punct de vedere diagnostic. Decizia de continuare sau de întrerupere a sarcinii aparţine părinţilor, atât timp cât este respectat cadrul legal. Pacienţii cu displazii scheletale neletale pot avea un intelect normal, în ciuda aspectului fizic, şi trebuie făcute eforturi pentru acceptarea şi integrarea acestor pacienţi în societate.Bibliografie
1. Krakow D. et al. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009.
2. Superti-Furga A et al. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. 2007.
3. Goncalves L.F. et al. Newer imaging modalities in the prenatal diagnosis of skeletal dysplasia. Ultrasound. Obstet. Gynecol. 2004.
4. Camera G. et al. Birth prevalence of skeletal dysplasias in the Italian multicentric monitoring system for birth defects. Prog. Clin. Biol. Res. 1982.
5. Waller D.K. et al. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am. J. Med. Genet. A. 2008.
6. James D. High Risk Pregnancy - Management Options, 4th Ed. Elsevier Saunders 2011.
7. Gill K.A. et al. Ultrasound in Obstetrics and Gynecology. A Practitioner’s Guide. Davies Publishing, Inc 2014.
8. Dounghue V. et al. Radiological Imaging of the Neonatal Chest. Springer 2002.
9. Lachman R.S. et al. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias. Philadelphia: Mosby Elsevier 2007.
10. Eames B.F. et al. Molecular ontogeny of the skeleton. Birth Defects. Res. C. Embryo. Today 2003.
11. Campbell J. The fetal femur/foot length ratio: a new parameter to assess dysplastic limb reduction. Obstet. Gynecol. 1988.
12. Krakow D. et al. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009.
13. Ngo C. et al. First-trimester ultrasound diagnosis of skeletal dysplasia associated with increased nuchal translucency thickness. Ultrasound. Obstet. Gynecol. 2007.
14. Schramm T. et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet. Gynecol. 2009.
15. Superti-Furga A. et al. Molecular-pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet 2001.
16. Unger S et al. Double heterozygosity for pseudoachondroplasia and spondyloepiphyseal dysplasia congenita. Am. J. Med. Genet. 2001.
17. Unger S. et al. A diagnostic approach to Skeletal dysplasias. Pediatric bone. Elsevier Science (USA). 2003.
18. Jeanty P. et al. The assessment of the fetus with a skeletal dysplasia - SonoWorld.
19. Leena Ala-Kokko, James Hyland, Kerry Kocher Brown, Xiangwen Chen-Deutsch, Hongbin Liu. Skeletal dysplasia core & extended NGS panel. Disponibil la: http://ctgt.net/panel/skeletal-dysplasia-core-extended-ngs-panel. Accesat [Apr 2017].
2. Superti-Furga A et al. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. 2007.
3. Goncalves L.F. et al. Newer imaging modalities in the prenatal diagnosis of skeletal dysplasia. Ultrasound. Obstet. Gynecol. 2004.
4. Camera G. et al. Birth prevalence of skeletal dysplasias in the Italian multicentric monitoring system for birth defects. Prog. Clin. Biol. Res. 1982.
5. Waller D.K. et al. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am. J. Med. Genet. A. 2008.
6. James D. High Risk Pregnancy - Management Options, 4th Ed. Elsevier Saunders 2011.
7. Gill K.A. et al. Ultrasound in Obstetrics and Gynecology. A Practitioner’s Guide. Davies Publishing, Inc 2014.
8. Dounghue V. et al. Radiological Imaging of the Neonatal Chest. Springer 2002.
9. Lachman R.S. et al. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias. Philadelphia: Mosby Elsevier 2007.
10. Eames B.F. et al. Molecular ontogeny of the skeleton. Birth Defects. Res. C. Embryo. Today 2003.
11. Campbell J. The fetal femur/foot length ratio: a new parameter to assess dysplastic limb reduction. Obstet. Gynecol. 1988.
12. Krakow D. et al. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009.
13. Ngo C. et al. First-trimester ultrasound diagnosis of skeletal dysplasia associated with increased nuchal translucency thickness. Ultrasound. Obstet. Gynecol. 2007.
14. Schramm T. et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet. Gynecol. 2009.
15. Superti-Furga A. et al. Molecular-pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet 2001.
16. Unger S et al. Double heterozygosity for pseudoachondroplasia and spondyloepiphyseal dysplasia congenita. Am. J. Med. Genet. 2001.
17. Unger S. et al. A diagnostic approach to Skeletal dysplasias. Pediatric bone. Elsevier Science (USA). 2003.
18. Jeanty P. et al. The assessment of the fetus with a skeletal dysplasia - SonoWorld.
19. Leena Ala-Kokko, James Hyland, Kerry Kocher Brown, Xiangwen Chen-Deutsch, Hongbin Liu. Skeletal dysplasia core & extended NGS panel. Disponibil la: http://ctgt.net/panel/skeletal-dysplasia-core-extended-ngs-panel. Accesat [Apr 2017].