Congenital cystic adenomatoid malformation (CCAM) is a rare fetal malformation of the lower respiratory tract. After the development and frequent use of the obstetric ultrasound, those lesions can be detected antenatally, which leads to a proper intrauterine and neonatal management. In this article, we present the case of a term newborn who was diagnosed with congenital cystic adenomatoid of the lower left lung lobe. In evolution, the lung cysts increased in size, occupying almost entirely the left lung. However, the clinical evolution of the newborn was favorable, which made possible the delay of the surgical intervention for an older age.
CASE REPORT
Malformaţia chistică adenomatoidă congenitală a plămânului – prezentare de caz
Congenital cystic adenomatoid malformation of the lung – case presentation
Adriana Tecuci,
Simona Vlădăreanu,
Prof. Dr. Radu Vlădăreanu,
Simona Popescu,
Diana Costache,
Ciprian Pop-Began
First published: 07 mai 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.2.1.2018.1651
Abstract
Rezumat
Malformaţia chistică adenomatoidă congenitală (CCAM) a plămânului este o malformaţie congenitală a căilor respiratorii inferioare, rar întâlnită la făt. Odată cu dezvoltarea şi folosirea tot mai des a ultrasonografiei în timpul sarcinii, aceste leziuni sunt diagnosticate încă din viaţa intrauterină, ceea ce determină un managment intrauterin şi neonatal tot mai bun. În acest articol, prezentăm cazul unui nou-născut la termen, diagnosticat cu malformaţie chistică adenomatoidă a lobului inferior stâng la vârsta de 23 de săptămâni de gestaţie. În evoluţie, chisturile pulmonare au crescut în dimensiuni, ocupând aproape în totalitate plămânul stâng. Cu toate acestea, evoluţia clinică a nou-născutului a fost favorabilă, ceea ce a făcut posibilă amânarea intervenţiei chirurgicale până la o vârstă mai mare.
Introducere
Malformaţiile congenitale pulmonare sunt reprezentate de malformaţiile chistice adenomatoide şi de sechestrarea bronhopumonară, ele reprezentând anomalii în dezvoltarea tractului respirator inferior. Deşi sunt rare, chisturile adenomatoide reprezintă cea mai comună leziune congenitală pulmonară.
Utilizarea ultrasonografiei de sarcină în întreaga lume a dus la o diagnosticare prenatală mai fidelă a acestei malformaţii. Astfel, datele din întreaga populaţie raportează o incidenţă a malformaţiilor chistice adenomatoidale congenitale (CCAM) de 1 la 8.300 până la 35.000 de nou-născuţi(3,4), ceea ce reprezintă 25% din malformaţiile congenitale ale plămânului şi 95% din leziunile congenitale ale plămânului. CCAM apare sporadic şi nu este asociată cu factori predispozanţi, cu ar fi rasa, vârsta maternă, sexul sau expunerea la diferite substanţe. Este raportată o incidenţă uşor mai crescută în rândul feţilor de sex masculin. Sechestrarea bronhopulmonară (BPS) este şi mai rar întâlnită, neexistând o incidenţă raportată în populaţie. Atât CCAM, cât şi schestrarea bronhopulmonară reprezintă anomalii apărute în timpul dezvoltării arborelui bronşic.
Dezvoltarea pulmonară a fost împărţită în cinci etape distincte, bazate pe modificările anatomice care au loc în viaţa intrauterină: etapa embrionară (3-7 săptămâni), pseudoglandulară (7-17 săptămâni), canaliculară (17-29 de săptămâni) şi alveolară (36 de săptămâni până la termen). CCAM se dezvoltă în etapa pseudoglangulară (7-17 săptămâni)(5).
CCAM este o leziune hamartomatoasă cu conţinut tisular pulmonar cu origini diferite(6). În sechestrarea bronhopulmonară, leziunea este formată din ţesut pulmonar nefuncţional care s-a separat de ţesutul pulmonar cu structură normală. Ambele leziuni au potenţial de malignizare. În plus, pot exista şi leziuni hibride care reprezintă o combinaţie între cele două. Din punct de vedere patogenetic, teoriile includ proliferarea anormală a ţesutului pulmonar, obstrucţia căilor respiratorii sau diplazia şi metaplazia ţesutului celular normal.
Pe baza descrierilor histologice, a punctului de origine a leziunii şi a nivelului embriologic, în prezent sunt descrise cinci tipuri de malformaţie chistică adenomatoidă congentală:
- Tipul 0 este cel mai rar întâlnit şi se dezvoltă la nivelul traheii şi al bronhiilor(7). Are prognostic nefavorabil, ducând frecvent la deces. Chisturile sunt de dimensiuni mici.
- Tipul 1 este cea mai comună formă. Apare în 50-70% din cazurile de CCAM(7) şi se dezvoltă la nivelul bronhiilor distale şi al bronşiolelor proximale. Este compus din unul sau mai multe chisturi cu dimensiuni de la 3 la 10 cm(7). Chisturile sunt căpuşite de ţesut epitelial cu celule ciliate pseudostratificate, iar între chisturi se găseşte frecvent ţesut celular cartilaginos. Pereţii chisturilor sunt subţiri. Poate să apară hidropsul.
- Tipul 2 apare la nivelul bronşiolelor terminale şi reprezintă 15-30% din cazurile de CCAM. În acest tip de CCAM, chisturile sunt de dimensiuni reduse (0,5-2 cm)(7) şi sunt dispuse în arii solide, uneori greu de diferenţiat de ţesuturile din jur. Sunt căptuşite în interior de celule cuboidale ciliate sau de epiteliu columnar, dar pot fi găsite şi elemente de la nivelul alveolelor. Acest tip asociază frecvent anomalii (aproximativ 60% din cazuri). Prognosticul este determinat de tipul de anomalii asociate şi de numărul organelor cuprinse.
- Datorită dimensiunilor mici ale chisturilor, tipul 3 este descris ecografic ca o masă solidă şi hiperecogenă(7). Este descris la 5-10% din cazuri, iar prognoosticul depinde de suprafaţa pe care se extinde.
- Tipul 4 este format din chisturi mari, chiar şi de 10 cm diametru(7), având potenţial mare de malignizare, în mod special blastom pleuropulmonar.
Din punct de vedere ecografic, de multe ori CCAM şi sechestrarea bronhopulmonară sunt greu de diferenţiat. În general, tipurile 1 şi 2 sunt mai uşor de distins, datorită aspectului macrochistic, pe când tipul 3 este foarte asemănător ecografic cu imaginea din BPS. De asemenea, cele două pot fi diferenţiate şi prin imaginile Doppler: CCAM are vascularizaţie din circulaţia pulmonară, pe când BPS are flux direct de la nivelul aortei.
Cu timpul, imaginea ecografică de la nivelul plămânului devine mai ecogenă, ceea ce va determina o vizualizare mai dificilă a leziunilor. Totuşi, malformaţia se poate asocia şi cu alte semne, precum polihidramnios, revărsat pleural şi hidrops.
Tipul 2 al CCAM este cel mai frecvent asociat şi cu alte anomalii în aproximativ 60% din cazuri(7). Astfel, se pot asocia malformaţii cardiace (trunchiul arterial comun, tetralogia Fallot), agenezie renală, atrezie gastrointestinală sau anomalii scheletale.
Descoperirea antenatală a chisturilor trebuie să ghideze obstetricianul către o investigare ecografică amănunţită a fătului. Ecocardiografia fetală trebuie efectuată pentru a evalua funcţia cardică şi posibilele malformaţii cardiace asociate(6). Nu se recomandă de rutină efectuarea cariotipului, însă acesta poate fi util în cazurile în care se asociază mai multe anomalii, putând ghida conduita terapeutică(6). În cazurile în care există un cariotip modificat sau apar mai multe malformaţii asociate, se poate decide împreună cu familia întreruperea sarcinii. În situaţia apariţiei hidropsului, se poate efectua intervenţia antenatală. Intervenţiile care se pot efectua sunt toracocenteza, şunt toraco-amniotic, ablaţia laser sau injectarea unor agenţi sclerozanţi la nivelul aterelor care vascularizează chisturile. În general, apariţia hidropsului se asociază cu un prognostic rezervat, mortalitatea fiind mare. Dacă nu există un chist dominant care să fie evacuat, vârsta gestaţională este sub 32 de săptămâni şi se asociază hidrops, atunci se poate lua în considerare intervenţia directă prin operaţie deschisă materno-fetală (toracotomia sau lobectomia fetală)(5). Cu toate acestea, prognosticul este rezevat.
Managementul neonatal depinde de vârsta de gestaţie, localizarea şi dimensiunile chisturilor. Din cauza riscului ca pacienţii să devină simtomatici şi a posibilităţii de malignizare, se recomandă ca intervenţia să fie efectuată în primele luni de viaţă. Dacă nou-născutul este stabil din punct respirator, nu se impune intervenţie chirurgicală de urgenţă; intervenţia chirurgicală poate fi amânată până la maximum 10 luni de viaţă. Studiile relatează o mortalitate crescută în rândul nou-născuţilor şi al sugarilor la care s-a intervenit în urgenţă. La nou-născuţii care nu au asociat hidrops în perioada sarcinii, evoluţia a fost favorabilă, cu o supravieţuire de peste 95%(8,9). La feţii cu hidrops care au beneficiat de toracocenteză în viaţa intrauterină, s-a raportat o rată de supravieţuire de 80%, cu o supravieţuire neonatală de 69%(10,11).
Conduita terapeutică este strâns legată de tipul şi dimensiunea malformaţiei. Pentru evaluarea acestor sarcini, este necesară o echipă multidisciplinară formată din medicul obstetrician, neonatolog şi chirurg pediatru, pentru un managment cât mai bun al acestor nou-născuţi.
Prezentare de caz
Prezentăm cazul unui nou-născut la termen (38 de săptămâni), de sex masculin, născut pe cale vaginală, prezentaţie craniană, GN: 3730 g, scor Apgar 7/8.
Din antecedentele heredocolaterale, reţinem că mama, de 26 de ani, III G, II P, a mai avut o sarcină care s-a încheiat printr-un avort terapeutic, la vârsta de 22 de săptămâni, pentru o malformaţie cardiacă incompatibilă cu viaţa.
Sarcina a fost atent monitorizată, iar la vârsta de 23 de săptămâni, la efectuare morfologiei, s-au evidenţiat la nivelul toracelui stâng macrochisturi, dintre care unul de dimensiuni mari. Această zonă de macrochisturi se dezvolta pe o zonă hiperecogenă, aspectul fiind sugestiv pentru malformaţie chistică andenomatoidă congenitală (figurile 1 şi 2). În cadrul ecografiilor efectuate, nu au fost evidenţiate alte anomalii asociate. De asemenea, în trimestrul al doilea s-a efectuat şi o amniocenteză pentru cariotipare, cariotipul fiind normal.
La naştere, nou-născutul prezintă tegumente cianotice, apnee, stetacustic MV diminuat în hemitoracele stâng, raluri crepitante bazal drept, tiraj subcostal, AV =120 b/min, fără alte modificări pe aparate şi sisteme. Necesită aspirarea secreţiilor din cavitatea bucală şi ventilaţie cu presiune pozitivă pe mască şi balon timp de 30 de secunde pentru reluarea respiraţiilor spontane.
Pe secţie, imediat postnatal, a prezentat minim sindrom funcţional respirator, cu polipnee (FR =60 resp./min), discret tiraj subcostal, Sat. O₂ =95%, stetacustic MV diminuat la nivelul toracelui stâng, zgomate cardiace ritmice, AV =130 b/min. Este plasat în punct de neutralitate termică şi se administrează O₂ sub cort cefalic.
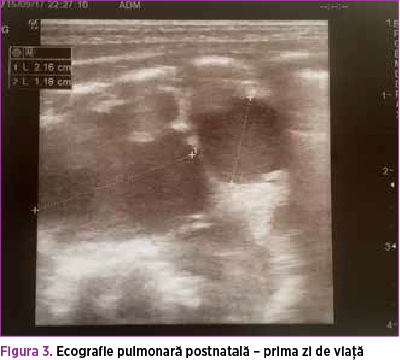
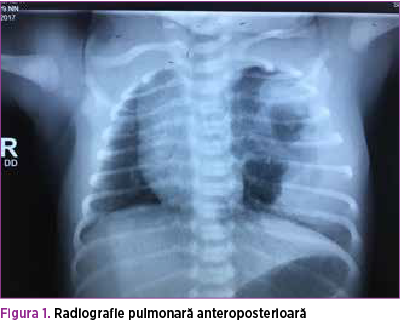
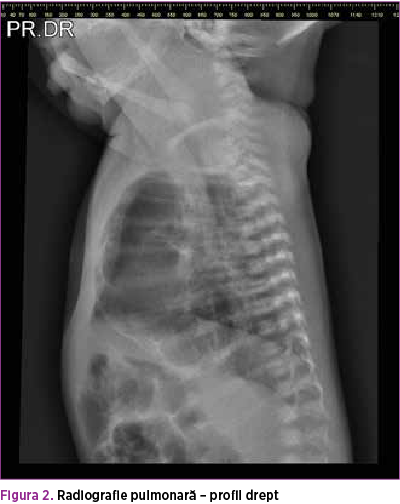
Se efectuează radiografie pulmonară (figurile 3 şi 4), care descrie neomogenitate a transparenţei pulmonare pe hemitoracele stâng prin distensie parţială aerică a parenchimului pulmonar şi prezenţa unei opacităţi mixte radiotransparente fără desen vascular şi bronşic şi a unei componente radioopace dispuse infero-lateral faţă de hemitorace. Transparenţă normală pe hemitoracele drept, cord cu volum normal. De asemenea, efectuarea ecografiei pulmonare (figura 5) confirmă prezenţa mai multor chisturi de diferite dimensiuni în hemitoracele stâng.
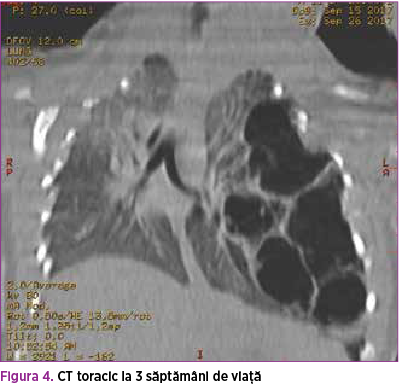
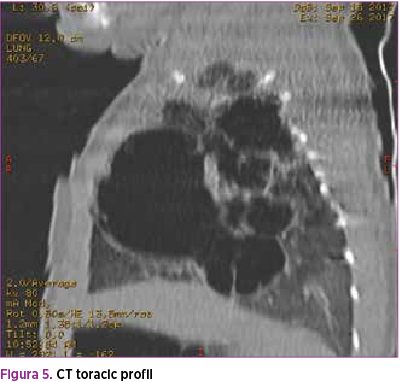
La 3 săptămâni de viaţă, CT-ul decelează la nivelul hemitoracelui stâng un conglomerat de leziuni ovalare cu pereţi subţiri, cu conţinut aeric, cea mai mare măsurând 75/60/31 mm (Dcr-cd/Dap/Dtr). Acestea ocupă aproape integral hemitoracele stâng, colabează segmentele pulmonare restante şi împing către dreapta cordul şi structurile mediastinale. Leziuni chistice pulmonare stângi sugestive pentru malformaţie adenomatoidă chistică congenitală (figurile 4 şi 5).
Paraclinic, gazele sangvine evidenţiază o uşoară acidoză respiratorie (pH=7,37; pCO₂=46 mmHg; pO₂=98 mmHg; HCO₃=22,9 mmol/L; BE=1,9 mmol/L). Restul evaluărilor paraclinice au fost în limite normale.
În evoluţie, s-a menţinut cu minim sindrom funcţional respirator, cu desaturări uşoare şi episoade de polipnee în timpul alimentaţiei şi în perioadele de agitaţie. Datorită evoluţiei clinice staţionare şi în urma efectuării consultului de chirurgie pediatrică, s-a decis temporizarea intervenţiei chirurgicale până în jurul vârstei de 6 luni. Copilul a fost evaluat periodic de un medic pediatru şi de chirurg, urmând ca în perioada următoare să se efectueze intervenţia chirurgicală.
Concluzii
CCAM este o afecţiune rară, cu prognostic favorabil dacă nu asociază hidrops în perioada perinatală. Progresia chisturilor pulmonare poate avea loc până la 28 de săptămâni, cu posibilitatea ca acestea să se reducă în dimensiuni în trimestrul al treilea de sarcină. Asocierea polihidramniosului sau a hidropsului are un prognostic nefavorabil, putând determina moarte fetală sau deces neonatal.
În cazul intervenţiilor materno-fetale în urgenţă, prognosticul neonatal este, de asemenea, rezervat. Se preferă temporizarea intervenţiei chirurgicale în defavoarea operaţiilor în urgenţă, atâta timp cât starea clinică a copilului o permite, acestea din urmă reprezentând un factor de risc major pentru creşterea mortalităţii neonatale.
Referitor la cazul prezentat mai sus, acesta se remarcă prin progresia chisturilor pulmonare, cu invadarea aproape în totalitate a parenchimului pulmonar stâng. Evoluţia clinică staţionară a permis temporizarea intervenţiei chirurgicale, pentru ca aceasta să fie efectuată cu un risc cât mai redus pentru pacient.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
1. Shanti CM, Klein MD. Cystic lung disease. Semin Pediatr Surg. 2008; 17:2.
2. Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg. 2014; 23:270.
3. Taguchi T, Suita S, Yamanouchi T, et al. Antenatal diagnosis and surgical management of congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther. 1995; 10:400.
4. El Amraoui W, Bentalha A, Hamri H, Es-Chrif El Kettani S, El Koraichi A. Congenital cystic adenomatoid malformation - dangers of misdiagnosis: a case report. J Med Case Rep. 2017;11(1):212.
5. Wilson DR, Hedrick HL, Liechty KW, Flake AW, Johnson MP, Bebbington M, Adzick NS. Cystic adenomatoid malformation of the lung: Review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet. 2006;140A:151-155.
6. Sfakianaki AK, Copel JA. Congenital Cystic Lesions of the Lung: Congenital Cystic Adenomatoid Malformation and Bronchopulmonary Sequestration. Rev Obstet Gynecol. 2012; 5:2.
7. Priest JR, Williams GM, Hill DA, et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44:14-30.
8. Witlox RS, Lopriore E, Oepkes D, Walther FJ. Neonatal outcome after prenatal interventions for congenital lung lesions. Early Hum Dev. 2011;87:611-618.
9. Stanton M, Njere I, Ade-Ajayi N, et al. Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg. 2009; 44:1027-1033.
10. Adzick NS, Harrison MR, Crombleholme TM, et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol. 1998; 179:884-889.
11. Cass DL, Olutoye OO, Cassady CI, et al. Prenatal diagnosis and outcome of fetal lung masses. J Pediatr Surg. 2011; 46:292-298.
2. Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg. 2014; 23:270.
3. Taguchi T, Suita S, Yamanouchi T, et al. Antenatal diagnosis and surgical management of congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther. 1995; 10:400.
4. El Amraoui W, Bentalha A, Hamri H, Es-Chrif El Kettani S, El Koraichi A. Congenital cystic adenomatoid malformation - dangers of misdiagnosis: a case report. J Med Case Rep. 2017;11(1):212.
5. Wilson DR, Hedrick HL, Liechty KW, Flake AW, Johnson MP, Bebbington M, Adzick NS. Cystic adenomatoid malformation of the lung: Review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet. 2006;140A:151-155.
6. Sfakianaki AK, Copel JA. Congenital Cystic Lesions of the Lung: Congenital Cystic Adenomatoid Malformation and Bronchopulmonary Sequestration. Rev Obstet Gynecol. 2012; 5:2.
7. Priest JR, Williams GM, Hill DA, et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44:14-30.
8. Witlox RS, Lopriore E, Oepkes D, Walther FJ. Neonatal outcome after prenatal interventions for congenital lung lesions. Early Hum Dev. 2011;87:611-618.
9. Stanton M, Njere I, Ade-Ajayi N, et al. Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg. 2009; 44:1027-1033.
10. Adzick NS, Harrison MR, Crombleholme TM, et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol. 1998; 179:884-889.
11. Cass DL, Olutoye OO, Cassady CI, et al. Prenatal diagnosis and outcome of fetal lung masses. J Pediatr Surg. 2011; 46:292-298.