From the clinician’s point of view, whenever there is an association between several malformations in the same patient, a syndrome must always be suspected. Furthermore, it has to be confirmed with the available diagnostic methods. Peters Plus syndrome is one of the rarest genetic syndromes; its prevalence is considered to be under 1:1000000, but the exact value remains unknown. About 100 patients diagnosed with Peters Plus syndrome have been reported in the literature. The syndrome is characterised by anterior chamber eye anomalies (central corneal clouding, iridocorneal adhesions), rhizomelia, typical facial features, developmental delay, congenital heart defects or structural brain malformations. The diagnosis can be confirmed by the identification of a B3GALTL mutation in a homozygous state (autosomal recessive inheritance). We present the case of a preterm infant, born at 35 weeks of gestation from a twin pregnancy obtained by in vitro fertilization, diagnosed with ocular Peters’ anomaly, congenital glaucoma, agenesis of the corpus callosum and coarctation of the aorta.
Malformaţii multiple la un prematur provenit din sarcină gemelară: sindrom Peters Plus? Prezentare de caz
Several malformations in a preterm infant from a twin pregnancy: Peters Plus syndrome? Case report
First published: 24 decembrie 2017
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.1.4.2017.1434
Abstract
Rezumat
În cazul asocierii de malformaţii la acelaşi pacient, clinicianul va suspecta întotdeauna un sindrom ce va trebui confirmat prin metodele diagnostice care îi stau la dispoziţie. Sindromul Peters Plus este unul dintre sindroamele genetice rar întâlnite, cu o prevalenţă considerată a fi sub 1:1000000 (cifra reală rămânând însă necunoscută). În literatura de specialitate sunt descrise aproximativ 100 de cazuri diagnosticate cu acest sindrom. Din punct de vedere clinic, sindromul se caracterizează prin modificări ale camerei anterioare a ochiului (leucom cornean, sinechii irido-corneene), rizomelie, facies carateristic, întârziere în dezvoltare, defecte cardiace sau neurologice. Diagnosticul de certitudine este genetic, iar defectul se află la nivelul genei B3GALTL, care suferă o mutaţie la nivelul ambelor alele (transmiterea fiind autozomal recesivă). Prezentăm cazul unui nou-născut prematur (35 de săptămâni), al doilea geamăn, provenit din sarcină obţinută prin fertilizare in vitro, ce prezintă anomalie Peters la nivel ocular, glaucom congenital, agenezie de corp calos şi coarctaţie de aortă.
Introducere
Sindromul Peters Plus a fost iniţial descris de Van Schooneveld în 1984(1), care a observat la 11 dintre pacienţi asocierea anomaliilor oculare ale camerei anterioare (anomalie Peters) cu statura mică, anomalii ale urechilor, cheilognatopalatoschizis şi întârziere în dezvoltarea neurologică. De asemenea, a raportat o rată crescută a avorturilor la mamele cu copiii afectaţi de acest sindrom(1,2).
În literatura de specialitate, acest sindrom se întâlneşte şi sub denumirea de sindrom Krause-Kivlin sau Krause-van Schooneveld-Kivlin şi cuprinde anomalii multiple ale diferitelor aparate şi sisteme, ce se asociază cu modificări ale camerei anterioare a ochiului. Cea mai frecventă dintre modificările oculare (dar nu şi patognomonică pentru acest sindrom) este anomalia Peters, ce include opacitate centrală corneană (leucom), subţierea regiunii posterioare a corneei şi aderenţe irido-corneene la periferia leucomului cornean. Se poate prezenta sub forma tipului I, cu afectare oculară uşoară, sau a tipul II, ce implică o afectare severă (cuprinde modificări ale cristalinului de tipul cataractei sau glaucomului congenital), asociat cu un prognostic nefavorabil. În practica medicală, tipul II este des întâlnit, iar afectarea este de cele mai multe ori bilaterală(3). Glaucomul congenital asociat cu anomalie Peters este mult mai dificil de tratat, comparativ cu glaucomul congenital primar. Atât tratamentul medicamentos, cât şi cel chirurgical reuşesc să normalizeze presiunea intraoculară la doar 32% din pacienţi, iar evoluţia ulterioară deseori este grefată de complicaţii postoperatorii sau de apariţia ambliopiei; astfel, prognosticul pe termen lung este infaust(4).
În cadrul sindromului sunt descrise modificări craniofaciale: aspect de „faţă rotundă” (pe parcursul copilăriei), frunte proeminentă, hipertelorism, filtrum lung, micrognaţie, aspect de arc al lui Cupidon al buzei superioare, urechi mici cu pedicul preauricular, cheiloschizis (45%), palatoschizis (33%), gât scurt şi îngust(5,6).
La 83% din cazuri este citată întârzierea în dezvoltare, cu grade variate de afectare (uşoară - 34%, moderată - 20%, severă - 26%). Frecvent, pacienţii prezintă statură mică, cu membre scurte, rizomelice; astfel, la vârsta adultă, pacienţii de sex feminin ajung să măsoare între 128 şi 155 cm, iar cei de sex masculin, între 141 şi 155 cm. Restricţia de creştere debutează prenatal, dar la naştere nu toţi pacienţii se situează sub percentila 3 pentru talie.
Defectele cardiace se întâlnesc în proporţie de 30% (frecvent se decelează defecte septale atriale, ventriculare, stenoză pulmonară ş.a.), iar printre anomaliile ocazionale se numără cele care afectează sistemul neurologic (agenezie/hipoplazie de corp calos, hipoplazie cerebeloasă, ventriculomegalie, hidrocefalie, mielomeningocel) şi aparatul reno-urinar(6).
În 2003, Heinonen et al.(7) descriu o nouă genă din familia glicozil-transferazelor, care, ulterior, în 2006, se dovedeşte a fi responsabilă de apariţia sindromului Peters Plus(8). Astfel, în zilele noastre, sindromul Peters Plus este definit ca o boală genetică transmisă autozomal recesiv, caracterizată prin mutaţii ale genei B3GALTL, situată pe braţul lung al cromozomului 13. Această genă codifică o proteină transmembranară, beta 1,3-galactozil transferază-like, implicată în procesul de O-glicozilare. Sindromul Peters Plus presupune mutaţii sau deleţii ale ambelor alele, ceea ce duce la pierderea funcţiei genei B3GALTL. În final, după translaţie, va rezulta o proteină trunchiată, cu un domeniu catalitic inactiv. Majoritatea pacienţilor diagnosticaţi la ora actuală cu acest sindrom prezintă mutaţii în stare homozigotă la nivelul unui situs de splicing (hot spot) în intronul 8 (c.660+1G>A)(5).
Este o boală rară, prevalenţa acestui sindrom considerându-se a fi sub 1/1000000, dar ea rămâne însă necunoscută. În întreaga lume, în literatura de specialitate, sunt citate aproximativ 100 de cazuri diagnosticate(9).
Prognosticul sindromului Peters Plus este variabil: se consideră, din punct de vedere al dezvoltării neuropsihomotorii, că toţi pacienţii vor dobândi achiziţii simple, inclusiv vorbirea, dar această dezvoltare va fi întârziată (80% din pacienţi). Totodată, sunt citate şi cazuri de adulţi cu funcţie cognitivă normală. Opacităţile corneene se pot diminua în primele 6 luni după naştere, dar niciodată corneea nu va suficient de clară pentru a permite o acuitate vizuală normală. Dimpotrivă, anomaliile severe ale camerei anterioare a ochiului pot duce la cataractă, respectiv glaucom, încă de la naştere. Cu toate acestea, modificările menţionate apar de obicei la distanţă de momentul naşterii. Având în vedere că retina şi nervul optic nu sunt afectate, se recomandă transplant de cornee la cazurile severe, la 3-6 luni de viaţă, pentru a preveni instalarea ambliopiei(5).
Prezentare de caz
Prezentăm cazul unui nou-născut prematur (35 de săptămâni), de sex feminin, al doilea geamăn, provenit din sarcină dispensarizată, gemelară, bicorială, biamniotică, obţinută prin fertilizare in vitro (FIV), extras prin operaţie cezariană (membrane intacte, lichid amniotic verde), prezentaţie pelviană, GN 2430 g (percentila 50 pentru VG de 35 de săptămâni), scor Apgar 9.
Menţionăm că a fost obţinut consimţământul informat al părinţilor pentru publicarea datelor nou-născutului în acest articol. Din antecedentele heredocolaterale reţinem că mama, de 47 de ani, a prezentat multiple sarcini pierdute, fiind diagnosticată cu trombofilie (heterozigotă pentru mutaţia genei MTHFR A 1298 C, heterozigotă pentru polimorfismul PAI-1 4G) şi a urmat tratament pe parcursul sarcinii cu acid acetilsalicilic 75 mg în primele 5 luni de sarcină şi heparină de greutate moleculară mică (enoxaparină) în lunile 6-8. Inseminarea s-a efectuat în 11.02.2017 (figura 1).

Sarcina a fost atent monitorizată, prin controale periodice lunare. Investigaţiile de laborator, atât înainte de inseminare, cât şi pe parcursul sarcinii, au fost în limite normale (analize de sânge, profil TORCH, culturi din colul uterin, uroculturi - fără modificări). La 23 de săptămâni de gestaţie, în urma evaluării ecografice - morfologie de trimestru II, geamănul II a fost diagnosticat cu agenezie de corp calos şi posibilă coarctaţie de arc aortic, în timp ce geamănul I nu prezenta modificări patologice.
La naştere, examenul clinic al geamănului II a evidenţiat la nivelul ochiului drept protruzie corneană, opacitate corneană a ambilor ochi, iar din punct de vedere cardiohemodinamic, AV>100 bătăi/minut, zgomote cardiace ritmice, suflu sistolic de grad III/VI în aria precordială, cu plus palpabil sincron la nivelul arterelor radiale şi diminuat, dar palpabil la nivelul arterelor femurale.
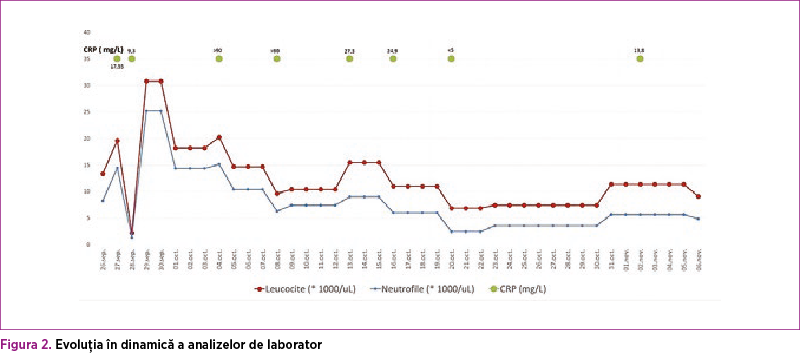
Pe secţie, imediat postnatal, geamănul II a prezentat detresă respiratorie care a necesitat intubaţie şi ventilaţie mecanică (în sistem Assist Control) timp de 5 zile. În evoluţie, la 48 de ore postnatal, a prezentat semne clinice şi paraclinice (leucopenie, procalcitonină pozitivă, ulterior leucocitoză cu neutrofilie, trombocitopenii repetate - figura 2) de sepsis precoce (confirmat de hemocultura pozitivă cu Klebsiella pneumoniae). A necesitat multiple scheme de antibioterapie cu spectru larg, conform antibiogramei (21 de zile), profilaxie antifungică, pentoxifilin, medicaţie vasopresoare, transfuzii multiple cu masă eritrocitară, concentrat trombocitar, plasmă proaspătă congelată şi imunoglobulină i.v., tratament sub care sepsisul a fost greu controlat.
Managementul acestui pacient a constat într-o abordare multidisciplinară, echipa fiind formată din medici neonatologi, pediatri, radiologi, cardiologi, oftalmologi, chirurgi cardiovasculari, medici de terapie intensivă şi geneticieni.
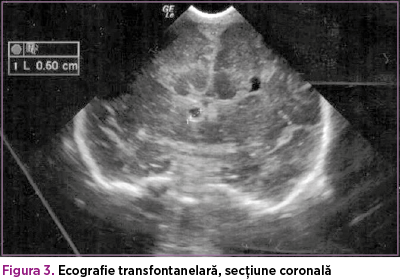
Din punct de vedere imagistic, ecografia transfontanelară efectuată postnatal a confirmat agenezia de corp calos şi a atestat existenţa unui chist în şanţul lenticulo-caudat (figurile 3 şi 4).
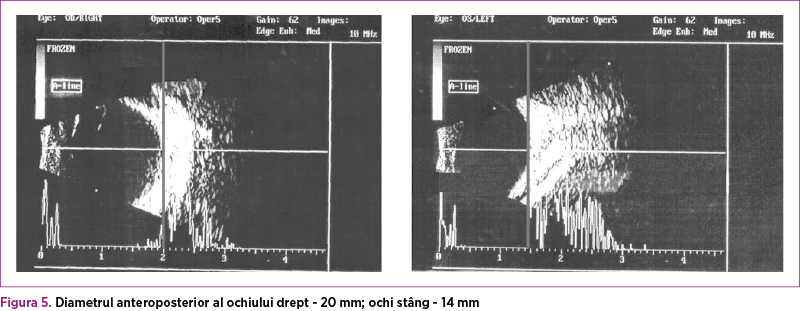
Consultul oftalmologic a evidenţiat la examinarea polului anterior - ochiul drept: buftalmie (consecinţă a glaucomului congenital), leucom cornean, cornee opalescentă, neovase corneene, ectazie corneană, iar la nivelul ochiului stâng: microftalmie, leucom cornean inelar şi cornee opalescentă (figura 5). La examinarea polului posterior, ambii ochi au prezentat vitros liber şi retina ataşată. În urma acestui consult, s-a ridicat suspiciunea de sindrom Peters (anomalii ale camerei anterioare oculare) prin prezenţa leucomului cornean bilateral, a sinechiilor iridocorneene şi a glaucomului congenital. La 20 de zile de viaţă, s-a intervenit chirurgical şi s-a efectuat trabeculectomie la nivelul ambilor ochi, iniţial cu evoluţie favorabilă.
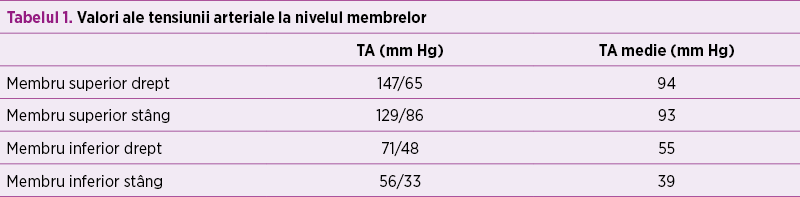
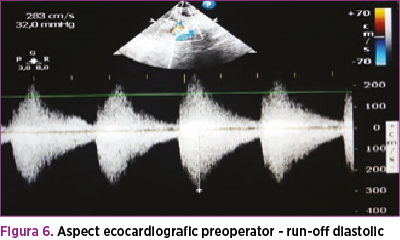
În paralel, s-a efectuat consult de cardiologie pediatrică, în urma căruia a fost confirmată coarctaţia de aortă, decelându-se aorta istmică, hipoplazică, de aproximativ 4 mm, ce se îngustează sub originea arterei subclavii stângi, la aproximativ 3 mm, cu viteza maximă la acest nivel 3 m/s şi run-off diastolic (figura 6); fracţia de ejecţie a fost iniţial de 60%, iar fluxul prin canalul arterial a fost 2 mm. Copilul a fost evaluat cardiologic periodic, iar la 4 săptămâni de la naştere, deşi sepsisul se considera rezolvat din punct de vedere medical, starea generală s-a degradat treptat, geamănul prezentând tegumente marmorate, timp de recolorare capilară de 3 secunde, transpiraţii profuze, accentuarea sindromului funcţional respirator cu desaturări de până la 80% în aerul atmosferic asociat, cu tensiune diferenţială semnificativă membre superioare - membre inferioare (tabelul 1), AV 180 bpm, suflu sistolic de grad IV/VI pe toata aria precordială, puls slab palpabil, bilateral, la arterele femurale (figura 7).
În urma reevaluării ecocardiografice, la acel moment s-a constatat FE a VS 35% şi flux turbulent imediat sub originea arterei subclavie stângi cu gradient maxim la acest nivel de 50 mm Hg, cu run-off diastolic.
S-a iniţiat PEV cu alprostadil 0,1 µg/kg/minut pentru a menţine deschis canalul arterial (la 3 zile după iniţierea tratamentului flux CAP 4,9 mm).
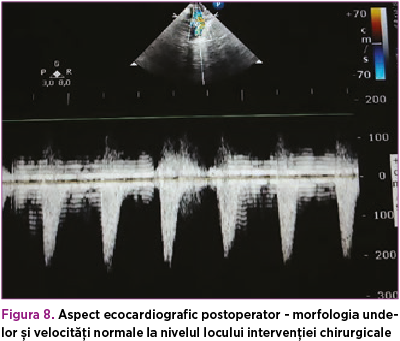
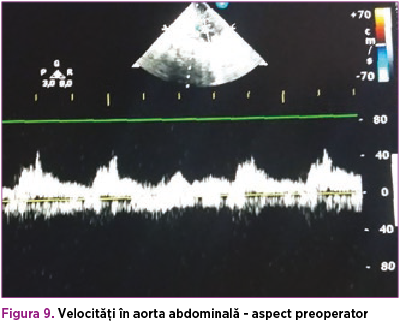
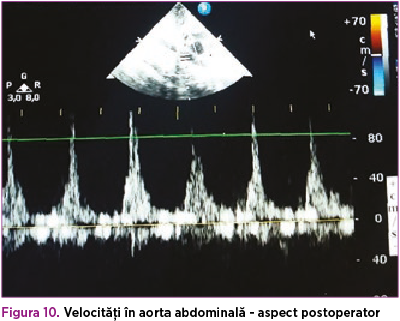
De asemenea, s-a suplimentat tratamentul cu dobutamină 5 µg/kg/minut, timp de 4 zile, pentru disfuncţia sistolică (FE 35%), ulterior aceasta din urmă s-a înlocuit cu adrenalină 0,05 µg/kg/minut, până la momentul operator. Astfel, la 42 de zile de viaţă, s-a efectuat corecţia chirurgicală a coarctaţiei de aortă, cu evoluţie postoperatorie favorabilă (figura 8). La domiciliu, copilul a primit tratament cu furosemid (1 mg/kg/zi) şi captopril (1 mg/kgc/zi) timp de o lună, cu sistarea ulterioară a terapiei şi fără restricţii din punct de vedere cardiologic (figurile 9 şi 10).
Pe de altă parte, evoluţia oftalmologică a fost nefavorabilă. În ciuda efectuării trabeculectomiei, diametrul anteroposterior şi presiunea intraoculară a globului ocular drept au continuat să crească; astfel, la 9 săptămâni postnatal, s-a efectuat evisceraţia globului ocular.
Discuţii
Având în vedere asocierea următoarelor malformaţii: anomalie Peters la nivel ocular, glaucom congenital, agenezie de corp calos, coarctaţie de aortă formă preductală (infantilă), s-a ridicat suspiciunea unui sindrom malformativ - sindrom Peters Plus.
Diagnosticul diferenţial s-a făcut cu anomalia Peters izolată, sindromul Cornelia de Lange, sindromul Smith-Lemli-Opitz, sindromul Robinow autozomal dominant, sindromul Rieger sau cu sindromul SHORT, majoritatea fiind infirmate prin manifestările fenotipice. S-a luat în discuţie şi sindromul de alcoolism fetal, care se poate asocia cu anomalie Peters, dar acesta a fost exclus în urma anamnezei mamei şi a faptului că nou-născutul provenea dintr-o sarcină gemelară, iar celălalt geamăn nu prezenta nicio modificare compatibilă cu expunerea la alcool pe parcursul dezvoltării intrauterine.
Pacienta a prezentat afectare oculară severă, compatibilă cu anomalie Peters de tipul II, la nivelul ochiului drept, care, în ciuda trabeculectomiei, nu a putut fi corectată. Presiunea intraoculară a ochiului drept a fost constant progresivă, fapt ce a condus la evisceraţia acestuia.
Faciesul, cât şi posibila existenţă a rizomeliei sunt dificil de analizat la nou-născut, neprezentând caracteristici marcante, cu excepţia patologiei oculare. Cu toate acestea, prin comparaţie cu geamănul I, geamănul II prezintă faţa rotundă, frunte proeminentă şi filtrum lung.
Deşi în literatură este citată drept componentă a sindromului, restricţia de creştere nu a fost prezentă la naştere - pacienta se afla la nivelul percentilei 30 pentru talie şi vârstă gestaţională. În primele 3 luni de viaţă nu a prezentat întârzieri în dezvoltare, dar considerăm că este încă precoce evaluarea prognostică din punct de vedere al dezvoltării.
Utilizând dotarea laboratorului de citologie/biologie moleculară, s-a efectuat testare genetică - cariotip clasic şi molecular (array CGH, folosind kitul 8 x 60 k Agilent), iar rezultatele obţinute au fost interpretate cu Agilent Cytogenomics Software, genome Assembly NCBI Build 37 (hg19), folosind baze de date internaţionale: DGV (Database of Genomics Variants), DECIPHER, OMIM (Online Mendelian Inheritance in Man) şi UCSC (University of California, Santa Cruz) Genome Browser. Analiza array CGH a identificat un profil normal feminin, fără modificări de tip microdeleţii sau microduplicaţii semnificative în regiunile cromozomiale analizate. Cu toate acestea, metoda are numeroase limite: nu identifică rearanjamente cromozomiale echilibrate, mozaicisme mici, triploidii, mutaţii punctiforme, dezechilibre genomice de dimensiuni mai mici decât rezoluţia sa, disomii uniparentale şi modificări epigenetice cum ar fi defectele de imprinting. Totodată, la o analiză a laboratoarelor acreditate pentru diagnosticarea sindromului Peters Plus, am constatat că există 59 de centre în Europa(9), dintre care doar şase folosesc ca metodă de diagnostic tehnica array. Dintre aceste şase centre, patru utilizează array CGH pentru a diagnostica acest sindrom, dar cu ajutorul unui kit cu rezoluţie superioară.
Având în vedere gradul ridicat de suspiciune clinică a acestui sindrom, nu putem exclude o posibilă diagnosticare ulterioară, utilizând fie alte tehnici, precum secvenţiere Sanger, NGS (Next Generation Sequencing), WES (Whole Exome Sequencing), fie tehnici bazate pe array (array CGH sau SNP array), dar cu utilizarea unui kit cu rezoluţie superioară.
Particularitatea acestui caz constă în asocierea la acelaşi pacient a trei malformaţii ce afectează sisteme şi organe diferite; cu toate acestea, diagnosticul de sindrom Peters Plus nu a putut fi confirmat. Totodată, se remarcă faptul că pacienta provine dintr-o sarcină gemelară; astfel, doar geamănul II a prezentat malformaţii, în timp ce geamănul I prezintă un examen clinic în limite normale. Un alt aspect compatibil cu posibilul sindrom Peters Plus ar fi existenţa numeroaselor sarcini pierdute în antecedentele mamei. Pe de altă parte, acest fapt poate fi explicat şi prin diagnosticul de trombofilie pe care aceasta îl poartă, reuşind să păstreze sarcina abia la vârsta de 47 de ani, după multiple avorturi spontane începând de la vârsta de 29 de ani.
Astfel, în cazul acestei paciente, considerăm că investigaţiile ulterioare trebuie să fie continuate. Prognosticul din punct de vedere neurologic şi cardiologic este relativ favorabil; sugarul este reevaluat periodic, fiind bine cunoscut riscul de restenozare la nivelul intervenţiei chirurgicale (anastomoză termino-terminală), mai ales în primul an postoperator. Din punct de vedere oftalmologic, se încearcă efectuarea, în viitor, a unui transplant de cornee la nivelul ochiului stâng pentru a asigura o acuitate vizuală cât mai bună şi, implicit, pentru a obţine maximul posibil privind calitatea vieţii pacientei.
Bibliografie
2 Hennekam RC1 et al: The Peters Plus syndrome: description of 16 patients and review of the literature. Clin Dysmorphol.; 2(4):283-300, 1993.
3. Zaidman et al. Long-term visual prognosis in children after corneal transplant surgery for Peters anomaly type I. Am J Ophthalmol, 144: 104-8; 2007.
4. Yang et al. Surgical management of glaucoma in infants and children`s with Peters’ anomaly: long term structural and functional outcome. Ophthalmology 111-112, 2004.
5. Kenneth Lyons Jones et al. Peters Plus syndrome, in Chapter T: Miscellaneous syndromes, p. 770-771, Smith’s recognizable patterns of human malformation, 7th edition, ed. Elsevier Saunders, 2013.
6. Saskia AJ Lesnik Oberstein et al. Peters Plus Syndrome GeneReviews®, https://www.ncbi.nlm.nih.gov/books/NBK1464/, accesat la 5.01.2018.
7. Heinonen TY, et al. A novel human glycosyltransferase: primary structure and characterization of the gene and transcripts. Biochem Biophys Res Commun. 309 (1): 166–174, 2003.
8. Lesnik Oberstein SA, Kriek M, White SJ, et al. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 79 (3): 562–566, 2006.
9. http://www.orpha.net/consor/cgi- bin/Disease_Search.php?lng=EN&data_id=968&Disease_Disease_Search_diseaseGro up=K&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseas es=Peters-plus-syndrome--Krause-van-Schooneveld-Kivlin-syndrome-&title=Peters- plus-syndrome--Krause-van-Schooneveld-Kivlin-syndrome- &search=Disease_Search_Simple, accesat la 5.01.2018
Articole din ediţiile anterioare
Artrogripoza la un nou-născut prematur provenit din sarcină gemelară. Prezentare de caz
Artrogripoza congenitală multiplă (AMC) este caracterizată în literatura de specialitate de multiple contracturi articulare congenitale la nou-născ...
Cauze non-placentare ale restricţiei de creştere uterină – studiu de caz şi review al literaturii
Restricţia de creştere intrauterină (RCIU) este definită ca eşecul unui făt de a-şi atinge potenţialul de creştere. Afectarea creşterii fetale se î...
Managementul durerii la nou-născut
Progresele efectuate în ultimii ani au stabilit că nou-născutul poate experimenta durere acută sau cronică, iar managementul corect are beneficii a...
Mortalitatea şi comorbiditatea la nou-născuţii transferaţi versus nou-născuţii din unitatea de grad 3
Mortality and co-morbidity in neonates transferred versus newborns from grade 3 unit