Displazia cleidocraniană – consideraţii clinice şi de diagnostic la doi fraţi
Cleidocranial dysplasia – clinical and diagnostic considerations in two brothers
Abstract
Cleidocranian dysplasia represents a genetic condition, which affects both genders, with autosomal dominant inheritance. Anomalies of the bones, associated with dental anomalies and small stature represent the clinical definitory elements that raised the suspicion of this condition. The authors present two clinical cases of two brothers who come from a family with similar anomalies.Keywords
cleidocranian dysplasiabone abnormalitiesfamilial aggregationRezumat
Displazia cleidocraniană este o boală genetică ce afectează ambele sexe, cu transmitere autozomal dominantă. Anomaliile osoase, asociate cu anomalii dentare şi statura mică au fost elemente clinice definitorii ce au ridicat suspiciunea asupra acestei boli. Autorii prezintă două cazuri clinice la doi fraţi care provin dintr-o familie în care există anomalii similare.Cuvinte Cheie
displazie cleidocranianăanomalii osoaseagregare familialăIntroducere
Displazia cleidocraniană (DCC), sau dizostoza cleidocraniană, cum a fost descrisă pentru prima dată de Pierre Marie şi Pierre Stainton în 1898, este o boală genetică ce afectează ambele sexe, cu transmitere autozomal dominantă(1,2). Prevalenţa este de 1 la 1 milionde persoane(4,8), cu penetranţă completă şi expresivitate variabilă. Boala este cauzată de o mutaţie la nivelul genei RUNX2 (core binding factor 1), care este localizată pe braţul scurt sau lung al cromozomului 6p21. Această genă codifică proteinele necesare pentru corecta funcţionare a osteoblastului şi este esenţială pentru diferenţierea celulelor dentare şi pentru formarea normală a oaselor(1-6). În majoritatea cazurilor, tulburarea de osificare este mai complexă, fiind afectată, pe lângă osificarea membranoasă, şi cea encondrală. Mineralizarea neregulată a metafizelor poate simula leziuni de condrodisplazie metafizară. Totuşi, 30-40% din cazuri apar spontan, aparent fără cauză genetică. Patogenic, apare o anomalie precoce de dezvoltare a ţesutului mezenchimal sau conjunctiv, cu osificare tardivă, sau, în unele cazuri, lipsă de osificare în anumite zone ale structurilor osoase(1,5).
DCC este adesea subdiagnosticată, poate pentru că, în comparaţie cu alte sindroame genetice, complicaţiile medicale sunt mai rare şi neinvalidante(4).
Scopul acestui articol este stabilirea unui diagnostic corect în cazul pacienţilor cu multiple anomalii osoase asociate cu statură mică.
Prezentarea cazurilor
Doi fraţi de sex masculin, în vârstă de 4 ani, respectiv 9 luni, se prezintă în serviciul de pediatrie pentru investigarea unui rahitism care nu răspunde la terapia corectă cu vitamina D.
Cazul 1 (fratele cel mare)
Istoric medical: primul copil, născut la 36 de săptămâni, prin operaţie cezariană, Gn=2700 g, scor APGAR=9, L=48 cm, perimetrul cranian=35 cm, perimetrul toracic=33 cm, alimentat natural un an, diversificat corespunzător, vaccinat corespunzător, cu dezvoltare neuromotorie şi cognitivă corespunzătoare vârstei.

Examenul clinic: greutatea=10,3 kg (-4DS pe scala Prader), talia=88,5 cm (-2DS).

Extremitatea cefalică: aspect de megacefalie, perimetrul cranian=51 cm (+2 DS), bose frontale, bose parietale, caput quadratrum, epicantus, hipertelorism, uşoară exoftalmie, baza nasului lăţită, buză superioară subţire, limbă în hartă geografică, boltă palatină ogivală, urechi discret coborâte şi rotate, fontanela anterioară deschisă=7/5 cm, normotensivă. Anomalii dentare cu malpoziţii şi dinţi supranumerari (figurile 1, 2).
Sistem osteoarticular: clinodactilie degetul 5 la mâini, genu valgum, picioare în talus valgus, mers „legănat”, torace îngust în partea superioară şi evazat la bază, aspect „în clopot”, stern uşor înfundat, poate apropia umerii pe linia anterioară, la mişcarea activă.
Aparat respirator, cardiovascular, digestiv în limite corespunzătoare vârstei, dezvoltare neuropsihică, motorie, cognitivă corespunzătoare.
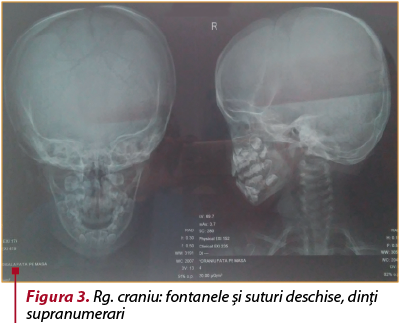
Examenele radiologice efectuate arată: craniu cu fontanele şi suturi deschise, dinţi supranumerari (figura 3), torace îngustat în partea superioară, evazat la baze, hipoplazie claviculară predominant dreaptă (figura 4), bazin displazic, disjuncţia simfizei pubiene (figura 5).


Cazul 2 (fratele mic)
Istoric medical: al doilea copil, născut la 32 de săptămâni, prin cezariană, Gn=2500 g, scor APGAR=8, L=47 cm, alimentat exclusiv natural până la 6 luni, diversificat corespunzător, vaccinat corespunzător vârstei, a primit mai multe doze injectabile de vitamina D, dezvoltare neuromotorie şi cognitivă corespunzătoare vârstei.
Examenul clinic: greutatea=6950 g (-2DS), talia=68 cm.
Extremitatea cefalică: perimetrul cranian=46,5 cm, fontane la anterioară=11/10cm, continuată cu suturi dehiscente posterioare, normotensivă, bose frontale, frunte bombată, bose parietale, circulaţie venoasă evidentă la nivelul scalpului, epicantus, hipertelorism, sclere albastre, absenţa dentiţiei.
Sistem osteoarticular: picior talus valgus bilateral, torace evazat la bază, umerii se pot apropia foarte mult pe linia mediană anterioară la mişcarea pasivă.
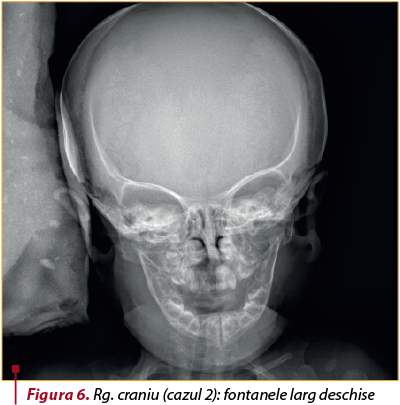
Examinările radiologice: fontanele larg deschise (figura 6), torace evazat la baze, clavicule hipoplazice, displazie de bazin, disjuncţia simfizei pubiene (figura 7).

Istoric familial
-
Mama: 36 de ani, greutatea=46 kg, talia=140,5 cm, fontanela anterioară deschisă=6,5/4 cm, cu dehiscenţa suturii posterioare, megacefalie, bose frontale, epicantus, hipertelorism, sclere albastre, întârziere în apariţia dentiţiei definitive, dinţi supranumerari, malpoziţionaţi (figura 8), clavicule hipoplazice (apropiere voluntară a umerilor pe linia mediană anterioară), genu valgum, picioare în talus valgus, fără fracture.
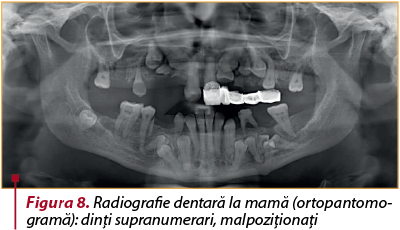
Figura 8. Radiografie dentară la mamă (ortopantomogramă): dinţi supranumerari, malpoziţionaţi -
Tatăl: 40 de ani, greutatea=52 kg, talia=158 cm, neagă boli cronice.
-
Bunica maternă: 60 de ani, greutatea=70 kg, talia=150 cm, fontanela anterioară închisă, apariţia dentiţiei definitive tardivă, poate apropia umerii la mişcarea voluntară spre anterior.
-
Mătuşa maternă: 28 de ani, greutatea=48 kg, talia=150 cm, fontanelă anterioară deschisă, erupţie dentară definitivă tardivă (la 18 ani), poate să îşi unească umerii pe linia anterioară; nu are copii.
-
Mătuşa maternă: 38 de ani, greutatea=45 kg, talia=135 cm, fontanela anterioară închisă, întârziere în apariţia dentiţiei primitive, poate apropia umerii la mişcarea voluntară spre anterior; fără copii.
Diagnostic diferenţial şi discuţii
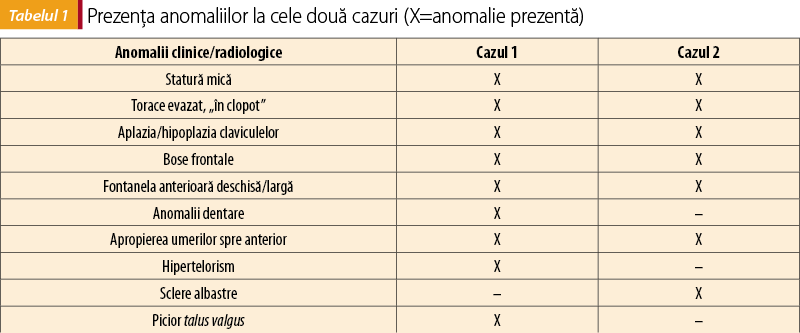
Existenţa unor anomalii similare la examenul clinic al celor doi copii, precum şi a unor aspecte similare la alţi membri ai familei sugerează ideea unei boli familiale sau a unui sindrom genetic. Hipermobilitatea umerilor, fontanele larg deschise, statura mică, anomaliile dentare, bose frontale şi hipertelorismul reprezintă indicii clare de DCC (tabelul 1).
Pentru confirmarea diagnosticului a fost efectuat şi examen genetic. Analiza cromozomilor prin cultura in vitro a limfocitelor T arată cariotip 46, XY; metafizele examinate nu au relevat prezenţa anomaliilor de număr sau de structură ale cromozomilor. Rezultatul citogenetic nu exclude prezenţa anomaliilor structurale nedecelabile prin această tehnică (microdeleţii, microduplicaţii sau translocaţii subtelomerice). Testul specific pentru gena RUNX2 (secvenţiere) nu a fost efectuat.
Pe baza istoricului familial, a examenului clinic şi a modificărilor imagistice, am concluzionat că suntem în faţa unor cazuri de displazie cleidocraniană. Diagnosticul pozitiv a fost stabilit pentru cei doi fraţi, mama lor, două mătuşi materne şi bunica maternă.
Clinic, în cadrul acestui sindrom se pot descrie: statură moderat redusă, inteligenţă normală, modificări scheletale ale extremităţii cefalice (craniu mare, „turtit”, frunte „bombată”, bose frontale şi parietale, suturi craniene închise târziu); masiv facial relativ mic, cu hipertelorism, rădăcina nasului lăţită şi adâncită, narine anteversate, boltă palatină ogivală, disodonţie; torace îngust în partea superioară, cu aplazia, hipoplazia sau displazia claviculelor, cu umerii „căzuţi” şi hipermobilitatea centurilor scapulare; bazin îngust; malformaţii vertebrale; membre extensibile, gracile, unghii hipoplazice, friabile. Radiologic se pot descrie: mineralizare întârziată a suturilor craniene şi a ramurilor pubiene, clavicule hipoplazice sau absente, pseudoepifizite ale metacarpienelor(7).
Pentru diagnosticul DCC, Mundlos a propus în 1999 o triadă de manifestări clinice pe care le consideră patognomonice, şi anume: dinţi supranumerari, absenţa completă sau parţială a claviculei şi suturi sagitale dehiscente sau închiderea tardivă a fontanelei sagitale(9).
Diagnosticul diferenţial se face cu alte entităţi care au caracteristici comune cu DCC.
Acondroplazia: boală genetică, transmisă autozomal-dominant, mutaţie în gena FGFR3(4q16.3), mutaţii de novo în multe cazuri (80-90%). Una dintre cele mai frecvente cauze de statură mică, disproporţionată, cu membre scurte, punte nazală aplatizată, cap mare, frunte bombată, degete scurte, în „trident”, lordoză lombară exprimată.
Osteogeneza imperfecta: transmitere autozomal-dominantă, gena mutantă 17q21.31-q32 nu asigură producerea normală de colagen tip I, mutaţii de novo.
Clinic, copiii prezintă fracturi produse de traume minore, laxitate ligamentară şi forţă musculară redusă. Vârsta de debut este variabilă (intrauterină, de la naştere până la vârsta de adult). Prezintă mare variabilitate clinică (6 tipuri clinice), cu malformaţii scheletice, statură mică, sclere albastre, dentinogeneză imperfectă. Radiologic, se decelează fracturi (subclinice). Rahitismul carenţial comun: boală metabolică produsă prin deficitul de vitamină D, care asociază semne osoase, hiperlaxitate ligamentară şi scăderea rezistenţei la infecţii. Paraclinic, se evidenţiază carenţa 25-(OH) vitaminei D, cu aspecte radiologice specifice.
Concluzii
Displazia cleidocraniană este o boală adesea subdiagnosticată, poate din cauza absenţei complicaţiilor medicale, în comparaţie cu alte sindroame. Diagnosticul poate fi dificil sau ignorat în cazurile fără anomalii morfologice evidente. Este esenţial istoricul familial pentru evaluarea şi încadrarea diagnostică. Diagnosticarea precoce este esenţială pentru abordarea terapeutică multidisciplinară, care necesită colaborare între pediatru, genetician, endocrinolog, chirurg maxilofacial şi ortodont. Prognosticul este bun, afectarea taliei este moderată, iar celelalte anomalii nu sunt invalidante.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Mundolos S. Cleidocranial dysplasia clinical and molecular genetics. J Med Genet. 1999; 36: 177-182.
- Bufalino A, Paranaíba LM, Gouvêa AF, Gueiros LA, Martelli-Júnior H, et al. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis. 2012; 18: 184-190.
- C N, Shakuntala BS, Mathew S, Krishnamurthy NH, Yumkham R. Cleidocranial dysplasia presenting with retained deciduous teeth in a 15-year-old girl: a case report. J Med Case Rep. 2012; 6: 25.
- Farronato G, Maspero C, Farronato D, Gioventù S. Orthodontictreatment in a patient with cleidocranial dysostosis. Angle Orthod. 2009; 79:178-185.
- Ott CE, Hein H, Lohan S, Hoogeboom J, Foulds N, et al. Microduplications upstream of MSX2 are associated with a phenocopy of cleidocranial dysplasia. J Med Genet. 2012; 49: 437-441.
- Suda N, Hamada T, Hattori M, Torii C, Kosaki K et al. Diversity of supernumerary tooth formation in siblings with cleidocranial dysplasia having identical mutation in RUNX2: possible involvement of nongenetic or epigenetic regulation. Orthod Craniofac Res. 2007; 10: 222-225.
- Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the South African experience. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013; 115: 46-55.
- Bhat MA, Laway BA, Mantoo S, Choudry K, Kotwal S, et al. Cleidocranial dysplasia: a rare cause of disproportionate severe short stature. Oman Med J. 2012; 27: 408-410.
- Diaconu G, Grigore I, Trandafir L, Anton D. Displazia cleidocraniană: consideraţii clinico-radiologice. Pract med. 2012; 27: 240-243.

Acizii graşi Omega-3, necesari pe tot parcursul vieţii
Studii recente, precum cercetarea realizată în 2018 la University of East Anglia, pun la îndoială rolul pe care acizii graşi Omega-3 îl au în reducerea riscului de boli cardiovasculare, unul dintre beneficiile pentru care medicii recomandă uleiul de peşte de decenii. Dezbatem acest subiect în Interviul...
Diabetul zaharat în sarcină – efecte asupra mamei şi copilului. Managementul în cabinetul medicului de familie
Ana-Aurelia Chiş-Şerban
În ultimii ani se înregistrează, la nivel mondial, dar şi în ţara noastră, o creştere alarmantă a vârstei de concepţie, a obezităţii şi a diabetului zaharat. Medicii de familie, cunoscându-şi pa...
Piciorul diabetic – complicaţie invalidantă a diabetului zaharat
Oana Manuela Spălăţelu, Sergiu Chirila, Leonard Gurgas, Vasile Sârbu
Piciorul diabetic reprezintă una dintre complicaţiile frecvente ale diabetului zaharat. Riscul pacienţilor diabetici de a face o ulceraţie sau de a necesita o amputaţie a devenit la ora actuală mai mare decât riscul de r...
Rezultatele sarcinii la femeile cu artrită psoriazică
Aida Petca, Cristina Mihai, Răzvan Petca, Florica Şandru, Claudia Mehedințu, Mihai Dumitraşcu, Ioana Cristina Rotar
Psoriazisul este o boală inflamatorie cronică, având consecinţe fizice şi psihologice importante. Acesta se caracterizează printr-un proces inflamator necontrolat şi un exces de citokine care provoacă disfuncţii endoteli...
Malformaţii congenitale cardiace frecvent întâlnite la copii
Adina Ungureanu, Corina Frecus, Sergiu Chirila
Malformaţiile congenitale cardiace reprezintă cele mai frecvente anomalii congenitale, fiind în acelaşi timp o cauză majoră de mortalitate infantilă. Studiul s-a desfăşurat pe o perioadă de doi ani, fiind analizate 74 de...