Sindrom de colestază neonatală la un prematur cu greutate foarte mică la naştere (VLBW) – prezentare de caz
Neonatal cholestasis syndrome in an very low birth weight (VLBW) premature – case presentation
Abstract
Cholestasis syndrome is one of the most challenging diagnoses in neonatal pathology (with an incidence of 1/2500 live births), especially in preterm babies. Biliary athresia is the most frequent cause of neonatal cholestasis (25-35%). Without an early diagnosis in the neonatal period and proper surgical intervention, it leads irreversibly to hepatic cirrhosis. The first approach in the management of neonatal cholestasis is to determine the level of direct bilirubin in every patient who associates prolonged jaundice (more than 14 days for term babies and 21 days for preterm babies). This paper presents a case study of a preterm baby with an early onset of neonatal E. coli sepsis complicated with neonatal cholestasis syndrome and modified results at the metabolic screening.Keywords
preterm babyneonatal cholestasisconjugated hyperbilirubinemiabiliary atresiasepsisRezumat
Sindromul de colestază neonatală reprezintă o patologie importantă (cu o incidenţă de 1/2500 de nou-născuţi vii), care ridică multe probleme de diagnostic etiologic în perioada neonatală, în special la prematuri(1). Atrezia biliară este cea mai importantă cauză de colestază neonatală (25-35%). În lipsa diagnosticării ei precoce în perioada neonatală, nu se poate interveni chirurgical la timp pentru a stopa evoluţia ireversibilă către ciroză hepatică. Primul pas în stabilirea diagnosticului de colestază îl reprezintă determinarea bilirubinei conjugate în cazul oricărui icter care durează mai mult de 14 zile de la naştere pentru nou-născutul la termen şi 21 de zile pentru prematur (cu sau fără scaun acolic). În această lucrare aducem în discuţie cazul unui prematur diagnosticat cu sepsis precoce cu E. coli, care a asociat în evoluţie sindrom de colestază şi valori modificate ale screeningului metabolic.Cuvinte Cheie
prematurcolestază neonatalăhiperbilirubinemie conjugatăatrezie biliarăsepsisIntroducere
Colestaza este caracterizată prin obstrucţia sau încetinirea fluxului biliar la orice nivel, de la hepatocit până la joncţiunea arborelui biliar extrahepatic cu duodenul, conducând la hiperbilirubinemie şi icter.
În perioada neonatală, primul pas în stabilirea diagnosticului de colestază este diferenţierea icterului cu bilirubină directă crescută (BD) faţă de cel cu bilirubină indirectă (BI)(3).
Hiperbilirubinemia conjugată este definită printr-un nivel seric al BD care depăşeşte 1 mg/dl când bilirubina totală serică (BTS) <5 mg/dl sau BD >20% din valoarea totală serică atunci când BTS >5 mg/dl(1,2). Hiperbilirubinemia directă nu este considerată niciodată fiziologică, deşi sunt autori care consideră anumite ictere colestatice la nou-născuţi fiziologice, datorită imaturităţii funcţionale excretorii(5).
Incidenţa colestazei neonatale este de 1/2500 de nou-născuţi vii(4). Importanţa stabilirii precoce a diagnosticului de colestază rezidă din posibilitatea apariţiei unor complicaţii redutabile (de exemplu, în atrezia biliară extrahepatică, în lipsa intervenţiei chirurgicale, în mai puţin de 60 de zile de la naştere se instalează ciroza)(4,6,7).
Etiopatogenie
Din punct de vedere etiopatogenic, sindromul colestatic neonatal (SCN) poate fi de origine intrahepatică sau extrahepatică.
Colestaza neonatală extrahepatică (CNE) are ca principală cauză atrezia biliară (25-35%)(1), iar colestaza neonatală intrahepatică (CNI) poate fi de natură infecţioasă, alloimună, metabolică/genetică sau toxică. Cele mai discutate etiologii ale CNI sunt: infecţiile virale (virus herpes simplex, cytomegalovirus, rubeola), bacteriene (Gram-pozitive, Gram-negative, în special Escherichia coli), parazitare (toxoplasmoză), sepsisul la nou-născuţii care primesc alimentaţie parenterală, defecte genetice (25%)(2) – sindromul Allagile, colestaza neonatală familială progresivă, fibroza chistică; defecte metabolice înnăscute (20%) – deficienţa de α1-antitripsină (10%), galactozemia, tirozinemia, tulburări ale metabolismului lipidic, tulburări mitocondriale, defecte de oxidare ale acizilor graşi; cauze toxice – nutriţie parenterală prelungită (peste 2-4 săptămâni) la prematurii extrem de mici sau la nou-născuţii cu sindrom de intestin scurt; de natură inflamatorie – sindromul hepatitei neonatale idiopatice (hepatita cu celule gigantice); restul de 30% fiind de cauză necunoscută(3) (figura 1).
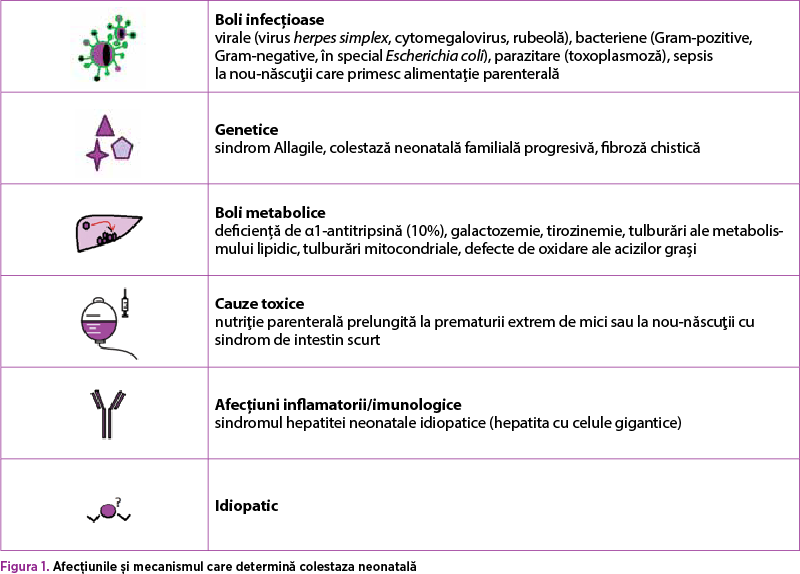
Principalele afecţiuni mai frecvente care duc la SCN
Atrezia de căi biliare extrahepatice (AB) (25-35%)(2) este o colangiopatie fibrozantă cauzată de obliterarea canalului hepatic comun sau a coledocului, la orice nivel de la vena portă până la duoden. Mecanismul fiziopatologic implică remodelarea incorectă a plăcii ductale în primul trimestru de viaţă intrauterină, asociată unor infecţii virale, mecanisme imunologice sau alterări ale sistemului vascular(8,9).
Sepsisul neonatal determină colestază (65,9%)(10) atât prin intermediul bacteriilor şi endotoxinelor bacteriene care duc la stază biliară, cât şi prin hemoliza pe care acestea o induc prin diverse mecanisme, inclusiv prin metode terapeutice (antibioterapia)(7).
Deficienţa de α1-antitripsină (10%)(2) este cea mai cunoscută cauză genetică de colestază neonatală cu transmitere autozomal recesivă. Mecanismul care stă la baza acestei patologii este reprezentat de o anomalie de plicaturare a proteinei α1-antitripsină şi secreţia inadecvată de către hepatocit a acesteia. Astfel, există un deficit sangvin, pulmonar şi o acumulare în ficat a α1-antitripsinei, care va determina colestază(11).
Sindromul Alagille (1-70000 NN)(11) este caracterizat de colestază asociată cu boli cardiace congenitale, facies dismorfic, vertebre „în fluture”, embriotoxon ocular posterior, anomalii renale, vasculare şi statură mică. Substratul morfopatologic al acestui sindrom este reprezentat de numărul mic al canaliculelor biliare intralobulare. Se transmite autozomal dominant şi are la bază mutaţia genei JAGGED 1, care codifică un ligand în calea de semnalizare Notch(11).
Colestaza asociată nutriţiei parenterale (28,2-49,8%)(13) are drept principală cauză administrarea emulsiilor lipidice, care prin conţinutul lor bogat în fitosteroli sunt toxice şi pot determina scăderea producţiei de bilă. Acestea au în compoziţie şi acizi Omega-3, care acţionează ca factori proinflamatori pentru ficat(15).
Colangita sclerozantă neonatală (<1/1000000)(14) se caracterizează prin inflamaţie şi fibroză obliterantă a canaliculelor biliare intra- şi extrahepatice, care alternează cu zone dilatate (segmente încă prezervate). Este o afecţiune autozomal recesivă, care se aseamănă histopatologic cu atrezia biliară(15).
Colestaza intrahepatică familială progresivă (1/50000-100000)(16) este un grup de cel puţin trei afecţiuni transmise autozomal recesiv, în care mutaţiile genelor implicate în formarea canaliculelor biliare duc la colestază progresivă şi injurie hepatocelulară(11).
Fiziopatologie
În colestază, mecanismul primar este cel al împiedicării excreţiei biliare, conducând la acumularea în exces în sânge a BD şi la scăderea sărurilor biliare din tractul gastrointestinal, ceea ce duce la malabsorbţia grăsimilor şi a vitaminelor liposolubile (A, D, E, K), în final rezultând o deficienţă vitaminică, nutriţională şi falimentul creşterii. În figura 2 este ilustrat metabolismului bilirubinei(6).
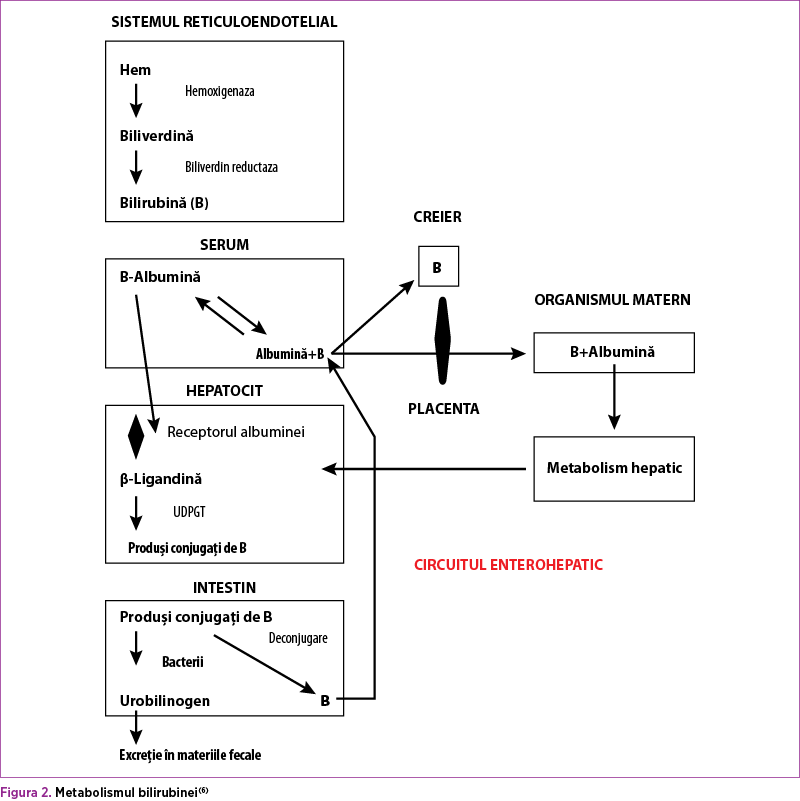
Diagnostic
Diagnosticul clinic. Semnele-cheie clinice sunt: icterul apărut tipic în primele două săptămâni de viaţă, cu nuanţă verdinică, asociat frecvent cu scaune acolice sau hipopigmentate şi urine hipercrome, hepatomegalie şi curbă ponderală nesatisfăcătoare. Scaunul acolic sugerează un proces obstructiv extrahepatic: atrezia biliară, chist coledocian sau dop mucos biliar. Când colestaza persistă, apare pruritul cronic şi falimentul creşterii, precum şi semne ale deficienţei vitaminice liposolubile. În afecţiunile care produc fibroză hepatică şi ciroză apar în stadii avansate semne şi simptome de insuficienţă hepatică acută sau ciroză precoce (hipertensiune portală, distensie abdominală prin ascită, dilatarea venelor abdominale, sângerări gastrointestinale din varicele esofagiene)(17,18).
Diagnosticul de laborator include(18):
- Bilirubina directă şi totală.
- Teste funcţionale hepatice: albumina serică, enzime hepatice – alanin aminotransferază (ALT) şi aspartat aminotransferază (AST), γ-glutamil transpeptidază (GGT), fosfatază alcalină (FA), probe de coagulare (PT/PTT, factorii II, VII, XI), amoniemie.
- Analize uzuale: glicemie, uree, creatinină, acid uric.
- Markeri infecţioşi: markeri serici de hepatită A, B, C, TORCH, culturi bacteriene/virale în urină şi scaun.
- Analize specifice pentru boli metabolice: serice – lactat, piruvat, 3-HO butirat, acetoacetat, acizi graşi liberi, acizi graşi cu lanţ foarte lung în anomalii peroxizomale, colesterol, trigliceride, fier, feritină, aminoacizi, carnitină totală şi liberă, α-fetoproteină, α1-antitripsină (activitate şi fenotip Pi), test genetic pentru galactozemie, transferină, free-T4 şi TSH în hipotiroidism, enzime lizozomale; din urină se evaluează aminoacizi, corpi cetonici, substanţe reducătoare/glucide (galactoză, fructoză), acizi organici, acizi biliari în anomalii de sinteză a acizilor biliari.
- Teste genetice pentru malformaţii sugestive trisomiilor 13 sau 18 – se efectuează cariotip.
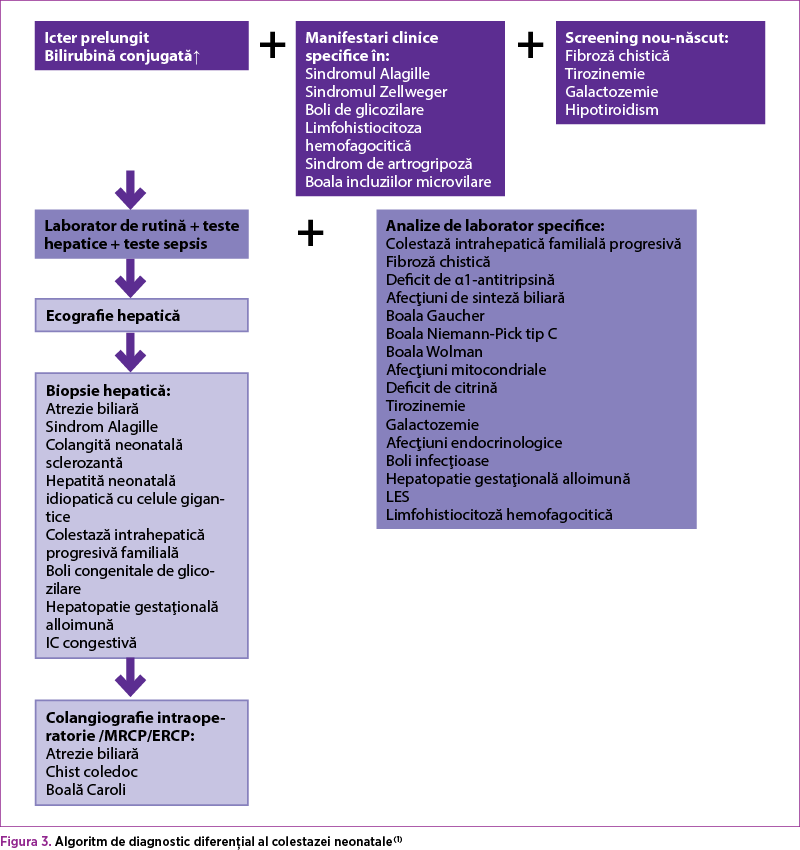
- După confirmarea hiperbilirubinemiei directe, se evaluează macroscopic scaunul. În obstrucţiile extrabiliare scaunele sunt acolice.
- Se prelevă probe de laborator de rutină sau specifice, când examenul clinic este sugestiv.
- Pentru diferenţierea precoce a atreziei biliare, a chistului coledocian şi a hepatitei neonatale se recurge la ecografie abdominală, puncţie biopsie hepatică şi scintigrafie hepatică.
- Ecografia abdominală este primul test imagistic de efectuat (în condiţii de repaus alimentar), care poate evalua dimensiunea şi structura hepatică, anumite anomalii ale vezicii biliare şi ale ductelor biliare comune (ductele biliare dilatate, chist coledocian, litiază biliară, sludge biliar şi specific AB – absenţa vezicii biliare şi semnul „triangular cord sign”)(1), lichid de ascită, evaluarea splinei (polisplenie prezentă în 100% din cazurile de atrezie biliară, splenomegalie prezentă în deficitul de α1-antitripsină, boala Wolman, tezaurismoze glicogenice etc.) sau malformaţii vasculare.
- Puncţia biopsie hepatică este considerată gold standard pentru diagnosticul atreziei biliare (90-95% din cazuri diagnosticate corect)(2) şi face diferenţierea de hepatita neonatală.
- Scintigrafia hepatobiliară este mai puţin specifică pentru diagnosticul AB (poate apărea şi în hepatita neonatală).
- În cazul în care investigaţiile anterioare sunt neconcludente, se recomandă colangiopancreatografie endoscopică retrogradă (ERCP)/rezonanţă magnetică (MRCP), scintigrafie/colangiografie intraoperatorie(1).
Management şi tratament
Având în vedere faptul că o mare parte dintre nou-născuţii cu colestază sunt subponderali şi au deficit nutriţional şi de vitamine liposolubile, principalul obiectiv este furnizarea unui conţinut caloric suficient pentru a compensa steatoreea şi pentru a preveni malnutriţia(19). Indiferent de etiologie, toţi pacienţii trebuie să primească formule de lapte praf şi suplimente cu vitamine liposolubile A, D, E şi K, precum şi trigliceride cu lanţ mediu (care se absorb din intestinul subţire fără ajutorul acizilor biliari)(9,20,21). În unele cazuri sunt necesare formule de lapte praf speciale (galactozemie, tirozinemie, fructozemie) (tabelul 1).
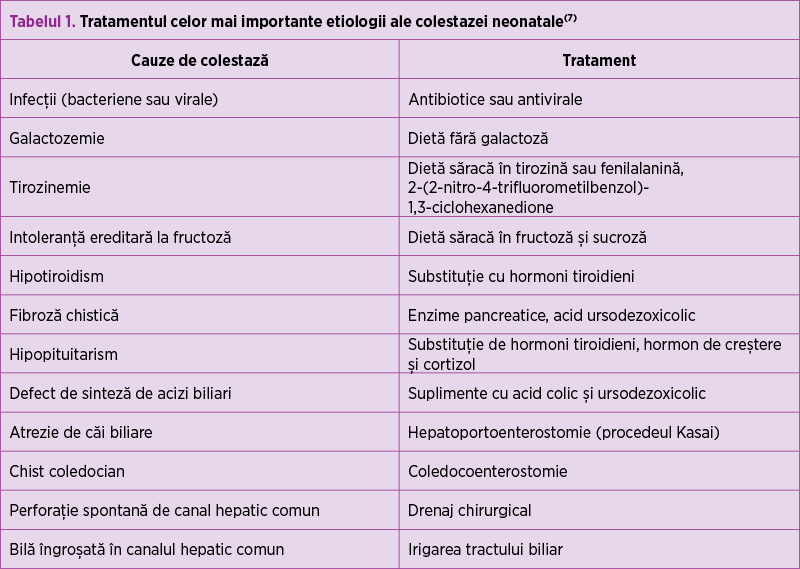
În cazul colestazei asociate sepsisului neonatal se indică tratament antiinfecţios empiric sau conform antibiogramei, iar în infecţiile congenitale (CMV, herpes simplex, Toxoplasma gondii etc.) este necesar tratamentul specific antiinfecţios(19).
Pentru combaterea pruritului se recomandă acidul ursodezoxicolic, rifampicina, colistiramina şi fenobarbitalul(19).
Se evită fototerapia la nou-născuţii cu colestază, din cauza apariţiei unei complicaţii rare – sindromul copilului bronzat, prin acumulare de porfirină şi alţi metaboliţi (clinic caracterizat prin coloraţie brun-cenuşie a tegumentelor, mucoaselor şi urinei). În aceste cazuri se recomandă exsangvinotransfuzia(7,8).
Tratamentul chirurgical include hepatoportoenterostomia Kasai pentru atrezia de căi biliare extrahepatice, care trebuie efectuată în primele 60 de zile. Chirurgia oferă rezultate excelente şi în cazul chisturilor coledociene.
Transplantul hepatic este terapia standard pentru ciroza decompensată de orice etiologie. La orice pacient căruia i s-a efectuat hepatoportoenterostomia Kasai, dar bilirubina rămâne peste 6 mg/dl la 3 luni după operaţie, trebuie considerat transplantul hepatic. Starea nou-născuţilor care se prezintă cu ciroză decompensată foarte probabil nu se va îmbunătăţi după hepatoportoenterostomie şi aceştia vor fi îndrumaţi direct spre un centrul de transplant hepatic(19).
Prognostic
Prognosticul colestazei depinde de cauza specifică şi este variabil, mergând de la o evoluţie benignă până la un curs progresiv către ciroză. AB este o afecţiune progresivă şi, dacă nu este tratată la timp, conduce la insuficienţă hepatică, ciroză şi hipertensiune portală în câteva luni de viaţă şi la deces până la un an de viaţă. Supravieţuirea după transplant hepatic în unele studii este de 70%(22). Hepatita neonatală idiopatică de obicei se rezolvă lent şi se poate complica cu afectare hepatică permanentă, insuficienţă hepatică şi deces(23).
Prezentarea cazului
Prezentăm cazul unui nou-născut prematur, de 31 de săptămâni (29 de săptămâni după aspectul morfologic), AGA (GN 1490 g – percentila 50), de sex feminin, provenit dintr-o sarcină cu risc (HTA preexistentă sarcinii), incomplet dispensarizată, extras prin operaţie cezariană (membrane intacte, contracţii uterine dureroase, lichid amniotic verde fluid), din prezentaţie transversă, indice Apgar 7 la 1 minut şi 8 la 5 minute. Din antecedentele heredo-colaterale reţinem: mama are 42 de ani, GV, PI, cu o sarcină extrauterină şi salpingectomie stângă în antecedente, HTA esenţială preexistentă sarcinii, în tratament cu Dopegyt, o capsulă la 6 ore, infecţie urinară cu E. coli tratată, dar nesterilizată, fără screening pentru TORCH, cezariană de urgenţă pentru preeclampsie (TAS maximă 210 mmHg). Din acest motiv nu s-a realizat profilaxia maturării pulmonare cu dexametazonă în cazul naşterii premature. La naştere, nou-născutul prezintă cianoză generalizată, echimoze multiple hemicorp stâng, AV peste100 bpm, iniţial respiraţii superficiale, urmate de respiraţii spontane, discret tiraj subcostal, tonus şi reactivitate scăzute. Pe secţia de TINN a necesitat ventilaţie noninvazivă cu PEEP (Neopuff® – PEEP 5 cm H2O, FiO2 40%, PIP 18 cm H2O), apoi în sistem CPAP nazal (PEEP 5 cm H2O, FiO2 40%), menţinut 24 de ore. S-a continuat cu administrare de O2 sub cort cefalic şi apoi în flux liber. Tratamentul medicamentos instituit a constat în: suport vasopresor cu dopamină 5 µg/kg/min, PEV de reechilibrare hidroelectrolitică administrate pe cateter venos central percutan, antibioterapie (ampicilină şi gentamicină).
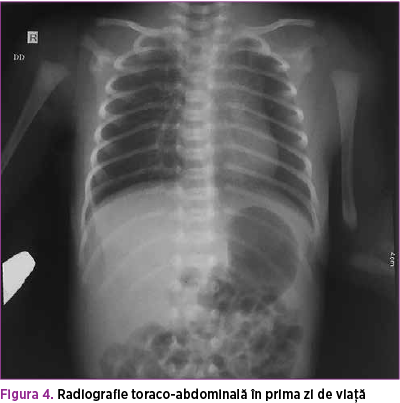
Sub tratamentul instituit se menţine echilibrat cardiorespirator, hemodinamic şi metabolic. Radiografia toraco-pulmonară iniţială arată infiltrat interstiţial difuz bilateral, mai evident în dreapta (aspect sugestiv pentru TTN sau pneumonie congenitală) – figura 4.
Din a doua zi de viaţă se evidenţiază icter pe fond eritrodermic şi se instituie fototerapia intermitentă. Paraclinic: BI 8,4 mg/dl, BD 0 mg/dl, CRP <5 mg/L, hemogramă şi EAB în limite normale. ETF normal la 3 zile de viaţă.
Evoluţia clinică este iniţial favorabilă; se menţine echilibrat cardiorespirator, se iniţiază alimentaţie enterală cu lapte matern pe SNG, cu toleranţă bună, urinează, elimină meconiu cu întârziere (la 4 zile de viaţă), reactivitate bună.
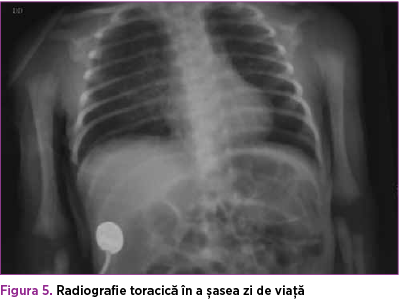
În ziua a cincea de viaţă, starea generală se modifică prin apariţia crizelor de apnee repetate, însoţite de bradicardie şi desaturare, acidoză respiratorie cu hipoxemie (EAB cu pH: 7,31, pCO2: 47 mmHg, pO2: 45 mmHg), accentuarea icterului neonatal. Se repune pe suport ventilator CPAP nazal (PEEP: 5 cm H2O, FiO2: 45%, PIP: 18 cm H2O) şi se adaugă aminofilin. În paralel cu alterarea stării generale, se modifică şi probele biologice, care devin sugestive pentru sepsis neonatal precoce cu E. coli (hemocultură pozitivă, sindrom inflamator prezent, CRP: 68 mg/L, trombocitopenie 47000/mmc); icterul se accentuează – BI: 14 mg/dl, BD: 0 mg. Se înlocuieşte antibioterapia iniţială cu meroponem, amikacină, vancomicină şi se completează cu fluconazol, pentoxifilin, albumină, masă trombocitară plasmă proaspăt congelată. Radiografia pulmonară evidenţiază desen pulmonar interstiţial accentuat supra-, peri- şi infrahilar de partea dreaptă şi discret accentuat perihilar stâng, sugestiv pentru pneumonie neonatală (figura 5). ETF-ul se menţine normal la 7 şi la 14 zile de viaţă.
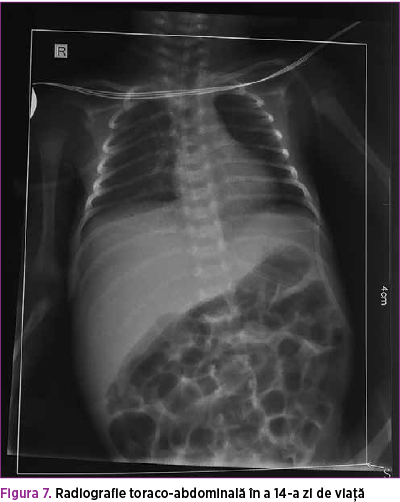
Evoluţia clinică ulterioară a fost nefavorabilă, cu agravarea continuă a stării generale: tegumente teroase icterice, TRC 4 secunde, sângerare la locurile de puncţie şi pe SNG, reapariţia crizelor de apnee sub CPAP şi aminofilin, abdomen moderat meteorizat, tranzit intestinal lent (elimină scaun de aspect meconial după microclisme evacuatorii), marginea anterioară a ficatului la 2 cm sub rebord, splina nepalpabilă, reactivitate uşor scăzută. Paraclinic s-a evidenţiat sindrom inflamator în creştere – CRP >90 mg/L, acidoză metabolică, leucocitoză cu deviere la stânga a formulei leucocitare, trombocitopenie severă, anemie (figura 6). S-a instituit tratament cu bolus SF, hemostatice, fc VII recombinant, ranitidină, albumină, PPC, MT, ME şi se modifică tratamentul antibiotic conform antibiogramei (ceftazidim + colistin + vancomicină), plus fluconazol. Se menţine alimentaţia enterală minimă trofică. Accentuarea detresei respiratorii impune intubarea orotraheală şi ventilaţia mecanică din ziua a noua de viaţă până în ziua a 12-a (PEEP 4 cmH2O, PIP 18 cmH2O, Fi O2 80%, FR 60 resp/min, Ti 0,36 s).
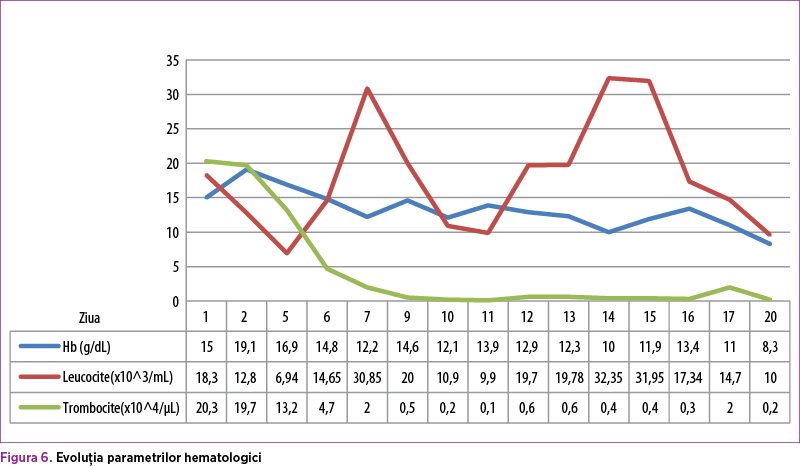
În prezenţa tratamentului intensiv instituit, evoluţia s-a menţinut severă, cu stare generală gravă şi apariţia icterului cu tentă verdinică din ziua a noua (BD 7,6 mg% la 13 zile de viaţă), abdomen moderat destins de gaze, suplu la palpare (figura 7).
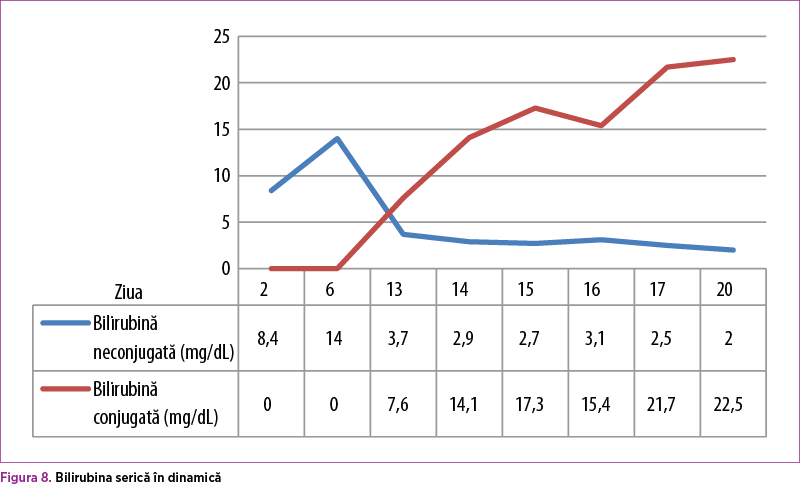
Sub tratament specific cu acid ursodezoxicolic şi protector hepatic, sindromul de colestază s-a accentuat progresiv, BD ajungând până la 22,5 mg/dl (figura 8). Ecografia abdominală şi consultul chirurgical elimină suspiciunea de obstrucţie de căi biliare externe, evidenţiind doar edem al peretelui colecistului (figura 9). Probele hepatice de citoliză sunt minim şi nespecific modificate: ALT menţinută normală, AST în creştere progresivă (36 – 44 – 58 – 88 U/L). GGT prezintă o valoare de 151 U/L în a 14-a zi, cu normalizare ulterioară – 44 U/L, colesterol total 12 mg/dl (valori scăzute), FA 389 U/L (normal), LDH 227 U/L (normal).
Screeningul metabolic extins efectuat pune în evidenţă niveluri semnificativ crescute de acyl-carnitină şi deficit de MCAD (medium chain Acyl-CoA-dehidrogenase) şi se recomandă testarea genetică şi consultul genetic. În ziua a 21-a de viaţă, după o evoluţie constant nefavorabilă, prezintă stop cardiorespirator, iniţial reversibil la manevrele de resuscitare.
Se efectuează ETF, care evidenţiază hemoragie intraventriculară masivă de grad IV. Ulterior, pacientul prezintă stop cardiorespirator ireversibil şi exitus.
Discuţii
În cazul prezentat, evoluţia clinică a fost marcată de semnele de sepsis neonatal cu debut din a cincia zi de viaţă, confirmat prin pozitivarea hemoculturii cu E. coli, care, în ciuda tratamentului instituit conform antibiogramei, nu s-au remis şi au condus în cele din urmă la exitus. Particularitatea cazului a constat în asocierea sindromului de colestază la patru zile de la debutul sepsisului. În literatura de specialitate se menţionează SCN ca o complicaţie neonatală cunoscută a infecţiei cu bacterii Gram-negative, în special E. coli(24). Rămâne în discuţie cauza decesului: sepsisul sau sindromul de colestază cu evoluţie nefavorabilă şi diagnostic neelucidat (doar prezumtiv). Se remarcă rezistenţa şi răspunsul nefavorabil la multiplele scheme de antibioterapie cu spectru larg, care ar putea fi explicate prin tratamentul iniţiat mamei în sarcină, în urma căruia infecţia urinară nu a fost sterilizată. Un alt factor de risc îl poate reprezenta rezistenţa scăzută la infecţii a prematurilor VLBW şi asocierea sindromului de colestază.
Având în vedere că în sarcină nu s-a efectuat screeningul pentru TORCH, pe lângă sepsisul cu E. coli, ar putea fi incriminate şi alte infecţii congenitale drept cauză de colestază. Nu au fost însă prezente în acest caz alte semne clinice caracteristice acestor infecţii, motiv pentru care nu s-au efectuat investigaţii specifice.
Cazul a prezentat, de asemenea, numeroase particularităţi clinice. Dintre semnele-cheie de colestază amintite anterior, au fost prezente: icterul cu nuanţă verdinică cu debut în a noua zi de viaţă, care s-a accentuat progresiv în ciuda tratamentului specific (acid ursodeoxicolic, protector hepatic); hepatomegalia apărută odată cu semnele de sepsis, urine tranzitorii hipercrome, falimentul creşterii. Pe de altă parte, nou-născutul nu a prezentat scaune acolice în niciun moment, doar tranzit intestinal lent; scaunele s-au menţinut cu aspect meconial, fapt ce exclude AB. De asemenea, şi ecografia a exclus AB şi alte malformaţii; nu s-a putut evalua imagistic colestaza intrahepatică.
În ceea ce priveşte afectarea hepatică, au fost prezente sângerări după ziua a şaptea de viaţă, odată cu accentuarea trombocitopeniei, iar paraclinic, probele hepatice au fost puţin şi nespecific modificate: GGT iniţial crescut, apoi normalizat; FA, LDH – normale; ALT normal; AST uşor crescut, dar nespecific şi colesterol scăzut; probele de coagulare nu s-au lucrat.
Un alt factor care ar fi putut influenţa metabolismul hepatic îl reprezintă medicaţia instituită, dintre care amintim: meropenemul, care se poate asocia cu disfuncţie hepatică, dar în acest caz a fost administrat doar 3 zile; fluconazolul cu administrare alternativă timp de 14 zile, posibil incriminat în apariţia icterului, şi ceftazidimul (administrat 8 zile), care poate determina creşterea valorilor LDH şi GGT.
Nutriţia parenterală poate fi luată în considerare în etiopatogenia SCN, în special la prematurii cu sepsis. În cazul nostru a fost administrată continuu, în paralel cu un aport enteral minim discontinuu.
Un factor benefic a fost însă neadministrarea de Intralipid®.
În urma screeningului metabolic extins s-au decelat niveluri semnificativ crescute de acyl-carnitină şi deficit de MCAD (medium chain acyl-CoA dehidrogenase).
În cazul primei tulburări metabolice sunt descrise hepatomegalie şi hipoglicemie, fiind cunoscut faptul că mulţi copii nu supravieţuiesc perioadei neonatale. Totodată, şi deficitul de MCAD se poate asocia cu hipoglicemie şi afectare hepatică.
Rămâne în discuţie dacă în condiţiile unui aport enteral insuficient, ale prematurităţii şi tratamentului, aceste valori pot fi concludente. Ar fi fost utilă repetarea screeningului extins.
Pentru elucidarea etiologiei ar fi fost necesar şi un consult genetic, precum şi efectuarea necropsiei, însă aceasta a fost refuzată de aparţinătorii legali.
Concluzii
Având în vedere istoricul antepartum, particularităţile clinice, paraclinice şi evoluţia în dinamică a cazului prezentat, putem interpreta drept cauze posibile de SCN sepsisul neonatal cu E. coli şi erorile înnăscute de metabolism (valori modificate ale acyl-carnitinei şi MCAD – medium chain acyl-CoA dehidrogenase).
Cu toate că valorile sunt semnificativ modificate, ele trebuie interpretate cu prudenţă, având în vedere interferenţele şi limitele acestui test la momentul recoltării. S-ar fi impus repetarea lor dacă nou-născutul ar fi supravieţuit.
În lumina celor discutate, diagnosticul cel mai probabil rămâne sepsisul neonatal cu debut precoce apărut la un prematur VLBW.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
1. Götze T, Blessing H, Grillhös C, Gerner P, Hoerning A. Neonatal cholestasis – differential diagnoses, current diagnostic procedures, and treatment. Frontiers in Pediatrics. June 2015;Volume 3, Article 43.
2. Feldman AG, Sokol RJ. Neonatal Cholestasis. Neoreviews. 14 November 2013.
3. Srivastava A. Optimizing Care and Outcome of Neonatal Cholestasis: Are We on the Right Track?. Indian J Pediatr 84. 2017;578–579.
4. Eichenwald E, Stark A, Hansen A, Martin C. Cloherty and Stark’s Manual of Neonatal Care. Wolters Kluwer. 2016; 22–32.
5. https://emedicine.medscape.com/article/927029-overview
6. Kaufman FR, Costin G, Thomas DW, Sinatra FR, Roe TF, Neustein HB. Neonatal cholestasis and hypopituitarism. Arch Dis Child 59. 1984;787–789.
7. Lane E, Murray KF. Neonatal Cholestasis. Pediatr Clin North Am 2017; 64. 621–639.
8. Dani C, Pratesi S, Raimondi F, Romagnoli C. Italian guidelines for the management and treatment of neonatal cholestasis. Ital J Pediatr 41. 2015;1–12.
9. Fischler B, Lamireau T. Cholestasis in the newborn and infant. Clin Res Hepatol Gastroenterol 38. 2014;263–267.
10. Bachtiar KS, Oswari H et al. Cholestasis sepsis at neonatology ward and neonatal Intensive Care Unit Cipto Mangunkusumo Hospital 2007: incidence, mortality rate and associated risk factors, Medical Journal of Indonesia. 2008; vol. 17, no 2.
11. Manuscript A, Cholestasis N. Neonatal Cholestasis 14. 2004;1–11.
12. https://ghr.nlm.nih.gov/condition/alagille-syndrome
13. Lauriti G, Zani A, Aufieri R, Cananzi M, Chiesa PL, Eaton S, Pierro A. Incidence, prevention, and treatment of parenteral nutrition-associated cholestasis and intestinal failure-associated liver disease in infants and children: a systematic review. JPEN J Parenter Enteral Nutr. 2014;Jan. 38(1):70-85.
14. http://www.orpha.net/consor/cgibin/Disease_Search.php?lng=EN&data_id=25219&MISSING%20CONTENT=Isolated-neonatal-sclerosing-cholangitis&search=Disease_Search_Simple&title=Isolated-neonatal-sclerosing-cholangitis.
15. Shetty NS, Shah I. Neonatal cholestasis due to primary sclerosing cholangitis. J Fam Med Prim Care 2016;5. 863–864.
16. https://emedicine.medscape.com/article/932794-overview
17. Gomella TL. Neonatology, 7th ed. McGraw Hill Education 2013; pg. 392-408, 663-685.
18. Popescu V. Actualităţi în pediatrie. Ed. Amaltea. 2008; vol 1.
19. Bhatia V. et al. Management of Neonatal Cholestasis: Consensus Statement of the Pediatric Gastroenterology Chapter of Indian Academy of Pediatrics. Indian Pediatr. 2014; Mar; 51(3):203-210.
20. Best C, Gourley GR. Management of neonatal cholestasis. Therapy 2009;6. 75–81.
21. Tufano M, Nicastro E, Giliberti P, Vegnente A, Raimondi F, Iorio R. Cholestasis in neonatal intensive care unit: Incidence, aetiology and management. Acta Paediatr Int J Paediatr 2009; 98. 1756–1761.
22. Lee WS, Chai PF, Boey CM, Looi LM. Aetiology and outcome of neonatal cholestasis in Malaysia. Singapore Med J. 2010; May, 51(5):434-9.
23. https://www.msdmanuals.com/professional/pediatrics/gastrointestinal-disorders-in-neonates-and-infants/neonatal-cholestasis.
24. Pereira NMD, Shah I. Neonatal cholestasis mimicking biliary atresia: Could it be urinary tract infection?. SAGE Journals. 2017; February 27.

Prognosticul materno-fetal şi managementul bolilor inflamatorii intestinale în sarcină
Andreea Boiangiu, Cristian Tiereanu, George Alexandru Filipescu, Cristina Maria Miron, Simona Vlădăreanu
Bolile inflamatorii intestinale (rectocolita ulcerohemoragică, boala Chron, colite neprecizate) afectează femeile tinere de vârstă fertilă, ducând la o provocare a obţinerii unei sarcini sănătoase. El...
Prognosticul materno-fetal al sarcinii complicate cu ciroză biliară secundară – review al literaturii de specialitate şi prezentare de caz
Andreea Boiangiu, George Alexandru Filipescu, Georgiana Dinu, Radu Vlădăreanu, Simona Vlădăreanu
Managementul gravidei cu afectare hepatică reprezintă o adevărată provocare pentru echipa complexă formată din obstetrician, gastr...
Malformaţia chistică adenomatoidă congenitală a plămânului – prezentare de caz
Adriana Tecuci, Simona Vlădăreanu, Radu Vlădăreanu, Simona Popescu, Diana Costache, Ciprian Pop-Began
Malformaţia chistică adenomatoidă congenitală (CCAM) a plămânului este o malformaţie congenitală a căilor respiratorii inferioare, rar întâlnită la făt. Odată cu dezvoltarea şi folosirea tot mai des...
Prognosticul materno-fetal al sarcinii complicate cu ciroză biliară secundară – review al literaturii de specialitate şi prezentare de caz
Andreea Boiangiu, George Alexandru Filipescu, Georgiana Dinu, Radu Vlădăreanu, Simona Vlădăreanu
Managementul gravidei cu afectare hepatică reprezintă o adevărată provocare pentru echipa complexă formată din obstetrician, gastr...
Malformaţia chistică adenomatoidă congenitală a plămânului – prezentare de caz
Adriana Tecuci, Simona Vlădăreanu, Radu Vlădăreanu, Simona Popescu, Diana Costache, Ciprian Pop-Began
Malformaţia chistică adenomatoidă congenitală (CCAM) a plămânului este o malformaţie congenitală a căilor respiratorii inferioare, rar întâlnită la făt. Odată cu dezvoltarea şi folosirea tot mai des...