Din aceeași categorie

Introduction
Wilson’s disease (WD) is an autosomal recessive copper metabolism disease involving the liver and/or the nervous system(1). In children, the main form is liver disease, with a very variable presentation, from mild elevation of transaminases to acute liver failure (ALF)(1,2). In adolescence, mainly in girls, WD can present as non-immune hemolytic anemia with ALF. The definition of acute liver failure in children is based on the PALF criteria (Paediatric Acute Liver Failure): no evidence of known chronic liver disease, hepatic-based coagulopathy that is not corrected by parenteral administration of vitamin K, international normalized ratio (INR) between 15 and 19.9 seconds in the present of the encephalopathy, respectively INR greater than or equal to 2 seconds, in the absence of encephalopathy(3-5). Acute liver failure in Wilson’s disease is a rare but highly aggressive form, with a high risk of death in the absence of liver transplantation(3). Early diagnosis and referral to a specialized center for the consideration of liver transplantation can be vital in these patients.
Case presentation
We report the case of a 13-year-old girl who was hospitalized in a regional hospital with abdominal pain and jaundice, with an evolution of three weeks. The patient was referred to our clinic for diagnosis and treatment after other causes of acute liver disease were excluded (acute viral infections or autoimmune hepatitis).
She did not have any history of hepatic disease, but her aunt died after an unspecified liver disease. She presented with jaundice, no sign of encephalopathy, with hemolytic anemia (hemoglobin level dropped in one day from 12 g/dl to 4.9 g/dl, with negative Coombs test), increase of transaminases (AST 164 U/L, low level of ALT – 8 U/L), very high bilirubin level (total bilirubin 35 mg/dl, conjugated bilirubin 32 mg/dl, with the increasing of total bilirubin level in one day up to 47 mg/dl), unmeasurable prothrombin time (over 120 s), unmeasurable INR. The ceruloplasmin level was 6 mg/dl, and the 24-hour urinary excretion of copper could not be dosed. Based on the clinical presentation and the laboratory parameters, the diagnosis was Wilson’s disease with non-immune hemolytic anemia and acute liver failure (Table 1). We administered chelating therapy with D-penicillamine and supportive therapy with fresh frozen plasma, antibiotics, glucose and arginine infusion, and erythrocytes transfusion. The modified Nazer score over 11 and the King’s criteria reveal the high mortality risk without liver transplantation. We referred the patient for emergency liver transplantation to the Fundeni Clinical Institute, Bucharest, which was successfully performed five days later. The genetic results confirmed the WD diagnosis homozygous status for ATP7B variant p.Lys844Ter (c.2530A>T).
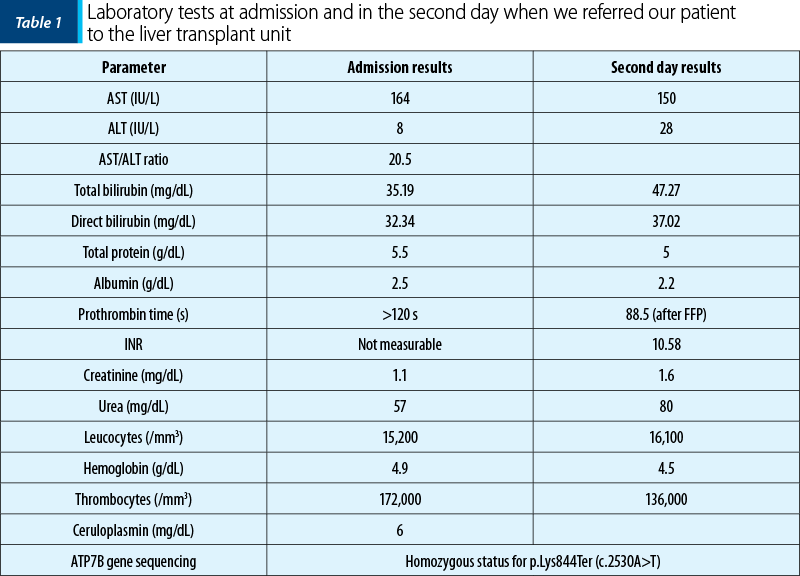
Discussion
We reported the case of an adolescent girl (13-year-old) who developed fulminant Wilson’s disease. She was admitted with severe acute liver failure, associated with non-immune hemolytic anemia and acute renal injury, requiring supportive therapy and emergency liver transplantation. The laboratory parameters were suggestive of WD, which was subsequently confirmed by genetic tests that detected the homozygous status for p.Lys844Ter (c.2530A>T) variant of the ATP7B gene. This is a rare variant of the ATP7B gene, and cases are described in the literature associated with the ALF form of WD.
Wilson’s disease is an autosomal recessive copper metabolism disease involving the liver, eyes, kidneys, nervous system, or other organs. The disease was described for the first time in 1912 by Kinnear Wilson as a chronic liver disease associated with progressive lenticular degeneration and neuropsychiatric symptoms(6). In the same year, Bernhard Kayser and Bruno Fleischer described the accumulation of copper in Descemet’s membrane (Kayser-Fleicher ring), and in 1993 the ATP7B gene was described(7). WD is a rare disease, with a prevalence estimated at 1 per 30,000-50,000 people in Europe(1,8). The gene ATP7B is located on chromosome 13 at the 13q14.3 locus. More than 600 different pathogenic variants have been identified(9). The ATP7B gene encodes the ATP7B protein, located in the hepatocytes, neuronal cells, erythrocytes, and renal cells. In the liver, ATP7B is responsible for intracellular copper transport and its incorporation with apocerulopasmin at the level of the Golgi complex(1,8,9). Thus, the ceruloplasmin forms and plays an essential role in plasma copper transport to the tissues where it is released and used(7,8). A large percentage (80%) of absorbed copper is biliary eliminated, the ceruloplasmin facilitating its passage into the biliary ducts. Variants of ATP7B impaired the processes of copper incorporation in ceruloplasmin and the excretion of the copper excess into bile(10). The result is the accumulation of copper in hepatocytes and later in other organs (central nervous system, cornea, kidneys, bones, or thyroid). The decreased ceruloplasmin serum level is due to the reduction of copper incorporation in hepatocytes and increased plasma copper(10).
In children, the main form of Wilson’s disease is represented by liver disease, with a very variable presentation, from mild elevation of transaminases to acute liver failure or cirrhosis with portal hypertension. In adolescence, mainly in girls, WD can be presented as non-immune hemolytic anemia with ALF, as it was in our patient. Few cases of children with WD developed ALF with hemolytic anemia(11). In acute liver failure, the massive destruction of the hepatocytes causes free copper release in circulation, inhibiting the erythrocyte’s glycolytic enzymes and determining acute hemolytic anemia(11). There are some differences between ALF in WD and ALF secondary to other causes. In Wilson’s disease, the level of transaminases (ALT 100-500 U/l) is low compared to the bilirubin level, which is very high (>20 mg/dl). Also, the alkaline phosphatase level is usually below 600 IU/L, and hemolysis is usually associated with a high level of copper in the blood, urine, or liver(9,12). At the renal level, APT7B protein is implicated in copper reabsorption at the level of the loops of Henle. In Wilson’s disease, ATP7B protein damage causes an increase in urinary copper excretion. In most cases with acute liver failure, the Kayser-Fleischer ring is missing(9,13).
The laboratory parameters in WD revealed low ceruloplasmin level (<20 mg/dl), increased urinary copper excretion (>5 µmol/24 h), or high levels of serum copper. All these parameters may have normal values, which does not exclude the Wilson’s disease(1,9).
The genetic test in WD with ALF presentation is limited, because there are many variants and due to the long duration for obtaining the results.
The most common variant in European populations is p.His1069Gln (c.3207A>G), with a prevalence of 30-70%, and it is predominantly encountered in young people. It is often associated with neurological and ophthalmic symptoms. The p.His1069Gln (c.3207A>G) mutation occurs when histidine from the N-domain SEHPL motif of ATP7B is replaced with glutamine acid, causing abnormal P-domain phosphorylation and decreasing ATP binding affinity(9). The hepatic form of the disease is associated with other variants, such as p.Arg778Leu (c.2333G>T), more frequent in some areas of Asia(14,15). These variants affect transmembrane copper transport and, as a result, like p.His1069Gln (c.3207A>G), they cause the decrease of ceruloplasmin level as a result of its increased degradation(16,17).
There are no clear genotype-phenotype correlations in Wilson’s disease(18). With a significant functional and structural impact on the ATP7B protein, the protein-truncating nonsense, frameshift, or splice-site variants may be associated with a more severe form of WD (early onset, low ceruloplasmin level, high copper content in the liver). In our cohort, the most frequent variants were p.His1069Gln (c.3207A>G), p.Gly1341Asp (c.4021G>A), p.Trp939Cys (c.2817G>T), and p.Lys844Ter (c.2530A>T). In patients presenting ALF associated with hemolytic anemia, p.Trp939Cys (c.2817G>T) and p.Lys844Ter (c.2530A>T) variants were more frequent than in other less severe forms, in which p.His1069Gln (c.3207A>G) was more frequent. Also, p.Gly1341Asp (c.4021G>A) had a similar frequency in all forms in our patients(19). In the reported patient, p.Lys844Ter (c.2530A>T) variant was demonstrated after a few weeks. In this variant, one nucleotide is deleted, and that causes a frameshift, predicting to result in a truncated protein. p.Lys844Ter (c.2530A>T) variant was described in WD patients of Hungarian origin and in a few patients with late onset of the disease(20,21).
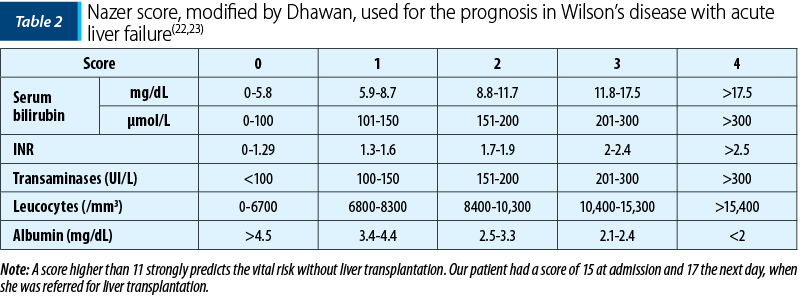
The prognosis in Wilson’s disease with acute liver failure is poor without the emergency liver transplantation. The indications for liver transplantation in children with WD are rare (<1%), and for a good selection of these cases, the Nazer score and its modified version are often used (Table 2). It is based on several laboratory parameters, such as bilirubin level, transaminases, albumin, INR and white blood cells. A score equal to or greater than 11 should be considered for liver transplantation because, otherwise, death will occur(1,9,13,22,23).
Conclusions
This case report aimed to raise awareness on the Wilson’s disease presentation as acute liver failure. The first step in the diagnosis is to remember this possibility as a cause of ALF. Acute liver failure in Wilson’s disease is a rare form of disease, with a high risk of death without liver transplantation. All teenagers presented with ALF, hemolytic anemia or neurological manifestations should be suspected of Wilson’s disease. Early diagnosis and referral to a specialized center for liver transplantation can be lifesaving. Also, chelating therapy should be started, and extracorporeal liver support could be used until the emergency liver transplantation is available.
Conflict of interest: none declared.
Financial support: none declared.
This work is permanently accessible online free of charge and published under the CC-BY licence.


Primul transplant hepatic din 2025 de la Spitalul Clinic Sfânta Maria
...
IRGH Cluj-Napoca a intrat în Programul Naţional de Transplant Hepatic
Institutul Regional de Gastroenterologie şi Hepatologie (IRGH) „Prof. Dr. Octavian Fodor” din Cluj-Napoca a intrat în Programul Naţional de Transplant Hepatic, iar primele intervenţii ar putea avea loc în luna mai a acestui an....