Condroblastomul osului: prezentare de caz, constatări imunohistochimice şi revizuirea literaturii
Chondroblastoma of bone: case presentation, immunohistochemistry findings and literature review
Abstract
Chondroblastoma is a rare benign cartilage-producing tumor, with an incidence of approximatively 1% of all primary bone tumors. It arises mostly in the second decade of life, affecting mainly the ends of the long tubular bones. We present two cases of chondroblastoma diagnosed and treated in the “Foişor” Orthopedics, Traumatology and Oesteoarticular TB Clinical Hospital, Bucharest, Romania. The purpose of this study is to contrast two different forms of presentation of the same histologic entity: one case of a 19-year-old male patient with a chondroblastoma located in the proximal humerus, and the case of a 37-year-old male patient with a chondroblastoma of the calcaneus. The morphological features, radiologic aspects and immunohistochemical studies have been assessed. None of the cases presented with specific radiologic signs for chondroblastoma, both lesions being osteolytic and well-delineated, in favor of a benign entity. The classic morphological aspects of chondroblastoma represented by tumor cells with round polygonal shape and nuclei with longitudinal grooves were associated with variable amounts of chondroid matrix, pericellular calcification and few randomly distributed osteoclast-like giant cells. The immunohistochemical studies showed the osteoblastic and chondroblastic phenotype of the neoplastic cells, sustained by positivity for CD56 and S100. DOG1, cytokeratin 8/18 and smooth muscle actin (SMA) were also found focally positive. All cases were treated by curettage with bone grafting, without recurrence in the short-term follow-up. Given the nonspecific radiologic findings and the histologic heterogeneity, chondroblastoma is sometimes misinterpreted as a malignant tumor, while this neoplastic lesion has a benign behavior and benefits from conservative surgical treatment only.Keywords
chondroblastomaprimary bone tumorcalcaneusproximal humeral epiphysisimmunohistochemistryRezumat
Condroblastomul este o tumoră benignă rară, având o incidenţă de aproximativ 1% din totalul cancerelor primare osoase. Se întâlneşte frecvent în a doua decadă a vieţii şi este localizat în majoritatea cazurilor la nivelul epifizelor oaselor lungi. Prezentăm două cazuri de condroblastom diagnosticate şi tratate la Spitalul Clinic de Ortopedie, Traumatologie şi TBC Osteoarticular „Foişor”, Bucureşti. Scopul acestui studiu este de a compara două forme diferite de prezentare ale aceleiaşi entităţi histopatologice. Primul caz este reprezentat de un pacient de gen masculin, în vârstă de 19 ani, cu condroblastom la nivelul epifizei humerale proximale, iar al doilea caz este al unui pacient de gen masculin, de 37 de ani, cu condroblastom localizat la nivelul calcaneului. S-au evaluat aspectele radiologice şi histologice ale tumorii şi s-a analizat profilul imunohistochimic al celulelor neoplazice. Investigaţiile imagistice au evidenţiat leziuni osteolitice bine delimitate, fără caractere de specificitate, cu aspect radiologic benign. Caracteristicile histopatologice clasice ale condroblastomului sunt reprezentate de celule rotunde, poligonale, cu nuclei indentaţi longitudinal, reniformi, asociate cu o cantitate variabilă de matrice condroidă, calcificări pericelulare şi rare celule gigante osteoclast-like distribuite aleatoriu. Testele imunohistochimice au demonstrat fenotipul osteoblastic şi condroblastic al celulelor tumorale, fiind pozitive pentru CD56 şi, respectiv, S100. De asemenea, DOG1, citokeratina 8/18, cât şi actina muşchiului neted (SMA) au fost pozitive focal. Ambele cazuri au fost tratate prin chiuretaj şi plombaj cu grefă osoasă, prezentând o evoluţie favorabilă, fără recidivă postoperatorie. Ţinând cont de aspectele radiologice nespecifice şi de heterogenitatea trăsăturilor histopatologice, condroblastomul poate fi perceput uneori ca o tumoră malignă, deşi această entitate are un comportament benign şi se tratează exclusiv chirurgical.Cuvinte Cheie
condroblastomtumoră primară osoasăcalcaneuepifiză humerală proximalăimunohistochimieBackground
Chondroblastoma is a rare benign cartilage-producing tumor, its incidence accounting for less than 1% of all primary bone tumors(1). Formerly known as “calcifying giant cell tumor” or “epiphyseal chondromatous giant cell tumor”, in 1942 the name of benign chondroblastoma of bone was introduced for the first time, describing its distinctive clinicopathologic features(2-3).
It arises mostly in the second decade of life, being more prevalent in men, and affects mainly the ends of the long tubular bones of the skeletally immature individuals. The common sites of involvement include the epiphysis of proximal humerus, proximal and distal femur and proximal tibia(3-6). In older individuals, the location varies more and may include non-tubular bones, craniofacial bones, bones of hands and feet(7). This entity has also been described in flat bones such as clavicle, sternum, ribs, vertebrae, pelvis and patella(5).
It is usually a solitary lesion, which involves the medullary cavity of the long bones(3). In flat bones, the tumor may extend into apophysis and into the articular space. Even though epiphysis of long tubular bones represents the common site of presentation, there are cases reported to arise in non-epiphyseal locations(8).
The most common clinical symptom is pain, which can be accompanied by other changes, such as swelling, stiffness, effusion and limitation in range of motion(6).
On plain radiographs, chondroblastoma appears as a well-circumscribed, eccentric, radiolucent lesion with a thin sclerotic rim(9). The computed tomography can reveal areas of mineralization. Calcifications appear as radiodensities and their amount varies greatly. When present in the chondroid matrix, they are described as arcs and rings of calcification(10). Adjacent cortex may be eroded or thinned, but rarely disrupted. In those cases associated with pathological fracture, a periosteal reaction may be seen. Lesions that do not arise in the epiphysis have nonspecific imaging findings(3).
The differential diagnosis based on radiologic imaging includes giant cell tumor of bone, enchondroma, chondromyxoid fibroma, low-grade chondrosarcoma, low-grade intramedullary osteosarcoma, and clear cell chondrosarcoma(11).
The peritumoral inflammatory reaction (especially edema) may be assessed through MRI studies(9). The intensity of T1- and T2-weighted MRI images depends on the amounts of various components within the lesion, such as cartilaginous component, cellularity, calcification, hemorrhage and cystic areas(9).
Tissue is often obtained from curettage and comes in many fragments, giving the impression of a lobular architecture. It presents as a grey yellow, friable material, usually with gritty cut surface calcifications. The chondroid component appears as a rubbery blue gray material, sometimes with bone consistency. The fragments may be hemorrhagic(3).
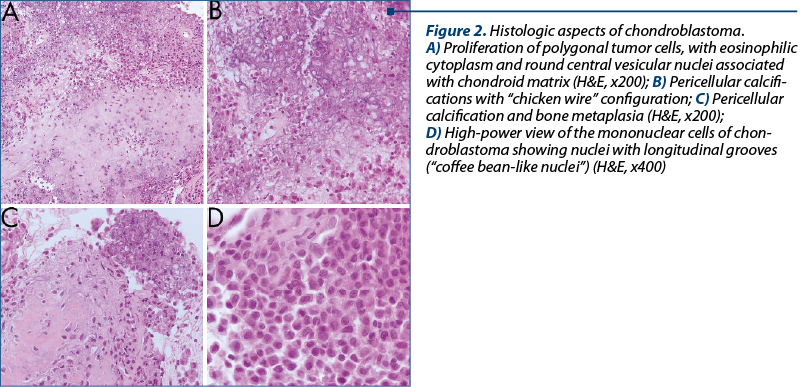
Histologically, this tumor is characterized by a proliferation of chondroblasts, chondroid matrix, calcifications, various amounts of giant cells, and is occasionally associated with aneurysmal bone cyst (ABC). Tumoral cells have a round polygonal shape, with well-defined cytoplasmic borders. The cytoplasm may be eosinophilic or focally clear. The nuclei have a characteristic appearance, with a central longitudinal groove (“coffee bean nucleus”)(3,12). The benign osteoclast-like giant cells of small dimensions are randomly distributed through the tumor, but their presence is not mandatory. Few mitoses may be visible, with a mitotic count of 1-3 mitoses/10 HPF. The presence of atypical mitoses excludes the diagnosis of chondroblastoma. Pericellular lace-like calcifications with “chicken-wire” appearance strongly support the diagnosis(3,11). Foci of hemosiderin deposition may be seen in the cytoplasm of the neoplastic cells. Half of the tumors of the hand and feet develop secondary ABC.
Some tumors may be locally aggressive, with cortical breakthrough and soft tissue invasion(11). Tumor necrosis and new bone formation can be seen when pathologic fracture is associated(11).
The immunohistochemical stains demonstrated that tumor cells are CD56, S100, CAM5.2 and vimentin positive. Focal positivity for keratins 8/18 and 19 was also described(13). An interesting relatively sensitive and specific marker for chondroblastoma is DOG1, which is known to be highly specific for gastrointestinal stromal tumors(14). SOX9 is positive in both chondroblastoma and chondromyxoid fibroma. Positive staining for Galectin-1 is considered reactive and is nonspecific, as it may be positive in 50% of chondroblastic osteosarcomas(10).
Recent studies identified a K36M mutation in H3 histone family member 3A (H3F3A) or 3B (H3F3B) genes, which proved a high sensitivity and specificity for chondroblastoma. Immunohistochemistry using antibodies against H3K36M is already available in some centers(15-18).
Depending on the histopathologic aspects, the differential diagnosis may include giant cell tumor of bone, chondromyxoid fibroma, secondary aneurysmal bone cyst, clear cell chondrosarcoma, and chondroblastoma-like variant of osteosarcoma(11).
The treatment of choice is the complete surgical curettage with or without bone grafting. As in giant cell tumor of bone (GCT), the chemical cauterization with phenol and cryotherapy may be used during the surgical procedure. En bloc resection may be necessary if aggressive recurrences occur and they cannot be treated by curettage(3-5). Amputation is an exceptional event.
Radiotherapy, previously used, is not yet recommended, as it leads to postradiation malignancy(3). It can be used when complete resection is not possible. Chemotherapy is not indicated(10).
The term “aggressive chondroblastoma”, which has no established relationship with the histologic appearance, was attributed to those exceptionally rare cases with lung metastases – more properly named lung implants – as they do not have the same unfavorable outcome as a malignant tumor(1,10).
Case presentation
We present two cases of chondroblastoma diagnosed and treated in the “Foişor” Orthopedics, Traumatology and Oesteoarticular TB Clinical Hospital, Bucharest, Romania. The patients were admitted for pain and movement limitation. Clinical and imagistic records, as well as treatment and postoperative evolution were gathered from the hospital record system. In our laboratory, we assessed histopathological diagnosis and immunohistochemistry (IHC) studies. Solid tumors diameters were measured using computed tomography.
Tumor tissue was fixed in 10% neutral buffered formalin and embedded in paraffin. The histologic sections were cut at 3 micrometers thickness. The standard stain was hematoxylin and eosin. Immunohistochemistry was carried out using LEICA BOND III automatic machine with Leica Bond antibodies for: CD 56 (clone 564), S100 polyclonal, SMA (clone osm1), CK8/18 (clone 5D3), and Ki-67 (clone MM1). DOG1 stain (Biocare mouse monoclonal antibody) was performed manually.
The histologic features evaluated included: giant cells, pattern of calcification, bone metaplasia, mitoses, chondroid material, cytologic atypia, necrosis, mature hyaline cartilage formation, and the presence of ABC.
Case 1
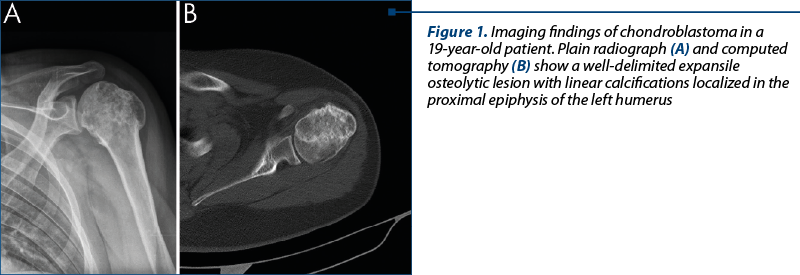
A 19-year-old male patient was admitted in our hospital for short-term persistent pain (two months) localized in the left shoulder. The imaging investigations revealed a well-delineated expansile tumoral lesion involving proximal left humeral epiphysis (Figure 1). The bone lesion measured 58.5/46/50 mm, was heterogenous, osteolytic, with increased peripheral bone density. As imaging findings favored a benign process, but associated with a rather broad differential diagnosis, a bone biopsy was performed.
The histopathology revealed a proliferation of polygonal tumoral cells, with eosinophilic cytoplasm and round central vesicular nuclei, some of which had longitudinal nuclear grooves (“coffee bean-like nuclei”). The sheet-like proliferation of tumoral cells was associated with chondroid matrix formation, bone metaplasia and focal calcifications of chondroid matrix (Figure 2). Pericellular calcifications with “chicken wire” configuration and tumor cells containing hemosiderin pigment were focally seen. Only a small number of osteoclast-like giant cells were present. Obvious cytologic atypia was absent.
After the diagnosis of chondroblastoma was rendered, the surgical treatment was decided. The patient underwent curettage and grafting with allograft bone. The postoperative course was uneventful.
Case 2
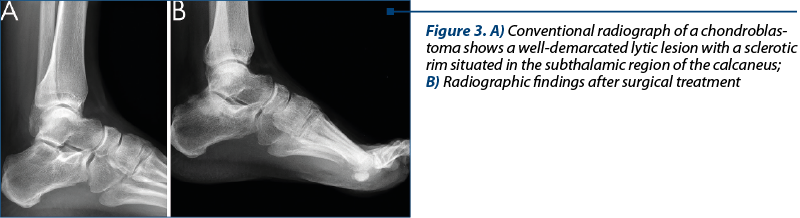
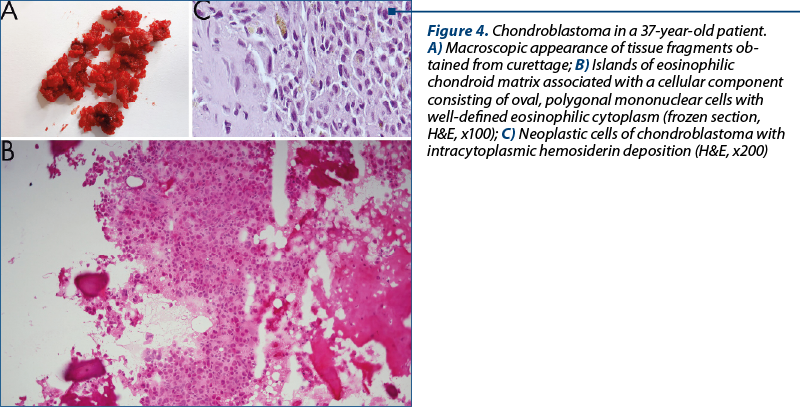
A 37-year-old male patient was admitted in our hospital for pain in the left foot. Conventional radiographs showed a well demarcated lytic lesion with a sclerotic rim, suspicious of a cystic benign lesion, situated in the subthalamic region of the calcaneus (Figure 3). Frozen section diagnosis of chondroblastoma was rendered (Figure 4B). The patient underwent definitive surgery in one step, with curettage and bone grafting with allograft bone.
The tissue obtained from curettage came in several fragments of cartilaginous consistency, with spotty calcifications, altogether not measuring more than 2/1 cm (Figure 4A). The microscopic features revealed sheets of polygonal cells with well-defined cell borders and nuclei with characteristic longitudinal grooves. These aspects were associated with islands of calcified chondroid matrix. “Chicken wire” pericellular calcifications were focally noticed. Rare osteoclast-like giant cells were randomly distributed. The mitotic activity was low, with no atypical figures. Hemorrhagic areas with hemosiderin deposition were also present (Figure 4C).
Both tumors were positive for CD56, DOG1, S100, CK 8/18, SMA and vimentin (Figure 5). In both cases, the Ki-67 proliferation index was very low: 1-3% of tumor cells.

Discussion
Chondroblastoma is a rare tumor of bone which can be easily diagnosed when classic morphological findings match the imaging data. Even though the radiological aspects may favor a benign diagnosis, several other entities could be envisaged in the differential diagnosis, among which some low-grade malignancies such as well-differentiated chondrosarcoma, low-grade intramedullary osteosarcoma, and giant cell tumor of bone.
The first case fits all the criteria described in literature, including the patient’s age and sex, the typical tumor location and the histopathological aspects. Even though conventional radiographs showed an osteolytic well-demarcated lesion in the humeral epiphysis, in favor of a benign process, malignancy could not be excluded on imaging findings. One particular feature was represented by bone metaplasia, which can mimic other tumoral entities, such as osteosarcoma, and the diagnosis may become really difficult on small biopsy material.
In the second case, the diagnosis of chondroblastoma was not anticipated, due to unusual clinical and topographic presentation, with radiological features consistent of a bone cyst. Chondroblastoma arising in adults tends to affect the short bones of the hand and feet, rather than the epiphysis of the long bones(19). Even though it was demonstrated that the histological aspects of this tumor in adults are prone to present with more calcifications, necrosis and other degenerative changes(19), we did not find necrosis in our specimen. Nevertheless, the histological features respected all the necessary criteria for the diagnosis of chondroblastoma, which was rendered on frozen sections and confirmed on paraffin.
Neoplastic cells with intracytoplasmic hemosiderin deposits were found in both of our tumors, this feature being more common among chondroblastoma of the craniofacial bones(3,10,11).
The histologic aspects that should raise concern are atypical mitoses, islands of mature cartilage in the tumor tissue, extensive necrosis and vascular, bone or soft tissue invasion. Neoplastic cells of chondroblastoma usually have a low number of mitoses, but only the presence of atypical mitoses should question the diagnosis of chondroblastoma(11). Our cases had a low mitotic index expressed through the proliferation index Ki-67 of 1-3% of the neoplastic cells.
If mature cartilage is present in the neoplastic lesion, the diagnosis of chondroblastoma should be carefully revised(10). This change may be apparent when a pathologic fracture is associated but, otherwise, an osteosarcoma or a chondrosarcoma should be taken into consideration. As a consequence, biopsy should not be taken from the fracture site, as this may lead to an erroneous diagnosis. Neither of these changes has been present in our cases.
As pulmonary implants have been described in several cases(1), our patients were also investigated with thoracic imaging, and neither of them had lung lesions. Soft tissue invasion and intraarticular extension may be seen in locally aggressive chondroblastoma or in long standing, neglected lesion, leading to radical surgery(12).
Depending on the various histopathologic aspects that can occur in a chondroblastoma, the differential diagnosis may include giant cell tumor of bone, chondromyxoid fibroma, secondary aneurysmal bone cyst, clear cell chondrosarcoma, and chondroblastoma-like variant of osteosarcoma.
-
Giant cell tumor (GCT) of bone usually occurs in skeletally mature bones. The lack of nuclear grooves in mononuclear cells, chondroid matrix and “chicken wire” calcifications are against the diagnosis of GCT. Moreover, giant cells tend to be evenly distributed among the tumor. The mononuclear cells are negative for S100, keratins and DOG1, and usually positive for p63(10).
-
Chondromyxoid fibroma may extend sometimes into the epiphyses, but lacks “chicken wire” calcifications and nuclear grooves, although both lesions can contain numerous osteoclast-like giant cells. It is characterized by centrally located hypocellular chondromyxoid matrix surrounded by hypercellularity. Immunohistochemistry shows a positivity for S100, but is negative for cytokeratins and DOG1(3,10).
-
ABC (especially solid type). It is preferable to do extensive sampling in order to identify tumoral tissue, when found centered in the epiphysis, because ABC can be a predominant finding and can overshadow an underlying chondroblastoma or other entity(10).
-
Clear cell chondrosarcoma should have invasive features and should contain cartilaginous matrix, which is absent in chondroblastoma(11).
-
Chondroblastoma-like variant of osteosarcoma presents an aggressive radiographic appearance, with tumor cells with greater degree of atypia than those found in chondroblastoma, and neoplastic osteoid(1,10,11).
Immunohistochemical studies are not always mandatory, but some markers still remain of interest in the controversial histogenesis of the tumor. DOG1 positivity is an unexplained finding in this type of tumor, this marker being highly expressed in gastrointestinal stromal tumor. DOG1 is also useful in the differential diagnosis with other giant cell rich tumors, its presence confirming the diagnosis of chondroblastoma(14). Cytokeratin positivity (CK8/18) is focally seen(10,14).
The intensity of the immunohistochemical stains varied depending on the markers used. The strongest expression was seen with CD56 (which is known as a surrogate for osteoblastic differentiation) and S100 (also strongly expressed in cartilaginous tumors), which was consistent with the dual accepted phenotype of the neoplastic cell, osteoblastic and chondroblastic (Figures 5A and 5B).
This polymorphic expression of immunohistochemistry markers highlights the unclear pathogenesis of this tumor. As chondroblastoma expresses genes involved in cartilage formation together with chondromyxoid fibroma, but also shares loss of heterozygosity pattern with low-grade chondrosarcoma, this tumor remains in the family of cartilaginous neoplasms(20,21). Transmission electron microscopy and immunoperoxidase studies also supported the cartilaginous origin of this tumor(3).
Recent studies identified a K36M mutation in H3 histone family member 3A (H3F3A) or 3B (H3F3B) genes, which proved a high sensitivity and specificity for chondroblastoma. A mutation in the H3F3B gene is more frequent. This mutation was not found in other histologic mimics such as GCT, GCT of Paget disease, ABC solid variant and osteosarcoma(18). Despite the fact that another mutation (G34W) in H3F3A gene was found in GCT of bone, this alteration was not present in chondroblastoma(15-18). Immunohistochemistry for K36M mutation is already available in some centers but, unfortunately, it is not available yet in our center, so we do not have any experience with it. We consider that its use may be beneficial in the differential diagnosis of giant cell-rich tumors and in confirming the morphological diagnosis of chondroblastoma.
The treatment of choice is curettage with bone grafting, and usually no further treatment is necessary. The recurrence rate in chondroblastoma was described to be around 14-18%(1), and was associated with tumor location or incomplete curettage(3,22). Some authors have reported an increased risk of recurrence in patients with chondroblastoma of flat bones or in those lesions associated with secondary ABC(3,23).
Both of our tumors were treated by curettage with bone grafting. One-step surgery, as done in the second case, is recommended when imaging data strongly favor a benign lesion. The diagnosis of chondroblastoma is confirmed by frozen section, thus reducing the patient’s stress regarding surgical procedures such as bone biopsy.
Conclusions
Osteoblastic and chondroblastic phenotype of the neoplastic cells in chondroblastoma is highlighted by the positivity for CD56 and S100, respectively. The polymorphic expression of IHC markers emphasizes the unclear histogenesis of the tumor.
Considering the radiologic findings and the histologic heterogeneity, chondroblastoma is sometimes misinterpreted as a malignant tumor. Its proper diagnosis is of outmost importance as this tumor has a benign behavior and benefits from conservative surgical treatment only.
Bibliografie
-
Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC Press; 2013; 24.
-
Jaffe HL, Lichtenstein L. Benign chondroblastoma of bone: a reinterpretation of the so-called calcifying or chondromatous giant cell tumor. Am J Pathol. 1942; 18:969-991.
-
Unni KK, Inwards CY. Dahlin’s Bone Tumors. 6th ed. Philadelphia: Lippincott Williams & Wilkins, a Wolters Kluwer Business. 2010.
-
Edel G, Ueda Y, Nakanishi J, et al. Chondroblastoma of bone: a clinical, radiological, light and immunohistochemical study. Virchows Arch A Pathol Anat. 1992; 421(4):355–366.
-
Kurt A-M, Unni KK, Sim FH, McLeod RA. Chondroblastoma of bone. Hum Pathol. 1989; 20(10):965–976.
-
Turcotte RE, Kurt A-M, Sim FH, Unni KK, McLeod RA. Chondroblastoma. Hum Pathol. 1993; 24(9):944–949.
-
John I, Inwards CY, Wenger DE, Williams DD, Fritchie KJ. Chondroblastomas presenting in adulthood: a study of 39 patients with emphasis on histological features and skeletal distribution. Histopathology. 2020; 76:308-317.
-
Brien EW, Mirra JM, Ippolito V. Chondroblastoma arising from a nonepiphyseal site. Skeletal Radiol. 1995; 24(3):220–222.
-
Shinmura K, Ishida T, Goto T, et al. Expression of cyclooxygenase-2 in chondroblastoma: immunohistochemical analysis with special emphasis on local inflammatory reaction. Virchows Arch. 2004; 444, 28–35.
-
Bocklage TJ, Quinn RH, Schmit BP, Verschraegen CF. Bone and soft tissue tumors. A multidisciplinary review with case presentations. London: JP Medical Ltd., 2014.
-
Nielsen GP, Rosenberg AE, Deshpande V, Hornicek FJ, Kattapuran SV, Rosenthal DI. Diagnostic Pathology: Bone, 2nd ed. Philadelphia: Elsevier, 2017.
-
de Silva MVC, Reid R. Chondroblastoma: varied histological appearance, potential diagnostic pitfalls, and clinicopathologic features associated with local recurrence. Ann Diagn Pathol. 2003; 7(4):205–213.
-
Bousdras K, O’Donnell P, Vujovic S, Henderson S, Boshoff C, Flanagan AM. Chondroblastomas but not chondromyxoid fibromas express cytokeratins: an unusual presentation of a chondroblastoma in the metaphyseal cortex of the tibia. Histopathology. 2007; 51: 414-416.
-
Akpalo H, Lange C, Zustin J. Discovered on gastrointestinal stromal tumour 1 (DOG1): useful immunohistochemical marker for diagnosing chondroblastoma. Histopathology. 2012; 60(7):1099–1106.
-
Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013; 45(12):1479–1482.
-
Amary F, Berisha F, Ye H, et al. H3F3A (Histone 3.3) G34W Immunohistochemistry: a reliable marker defining benign and malignant giant cell tumor of bone. Am J Surg Pathol. 2017; 41:1059-1068.
-
Amary MF, Berisha F, Mozela R, et al. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology. 2016; 69:121-127.
-
Schaefer IM, Fletcher JA, Nielsen GP, Shih AR, Ferrone ML, Hornick JL, Qian X. Immunohistochemistry for histone H3G34W and H3K36M is highly specific for giant cell tumor of bone and chondroblastoma, respectively, in FNA and core needle biopsy. Cancer Cytopathol. 2018 Aug; 126(8):552-566.
-
Negri S, et al. Clinicopathologic Analysis of Chondroblastoma in Adults: A Single-Institution Case Series. International Journal of Surgical Pathology. June 2020; https://doi.org/10.1177/1066896920927794
-
Romeo S, Oosting J, Rozeman LB, et al. The role of noncartilage-specific molecules in differentiation of cartilaginous tumors. Lessons from chondroblastoma and chondromyxoid fibroma. Cancer. 2007; 110:385–94.
-
Papachristou DJ, Goodman MA, Cieply K, Hunt JL, Rao UN. Comparison of allelic losses in chondroblastoma and primary chondrosarcoma of bone and correlation with fluorescence in situ hybridization analysis. Hum Pathol. 2006; 37(7):890-898.
-
Springfield DS, Capanna R, Gherlinzoni F, Picci P, Campanacci M. Chondroblastoma: a review of seventy cases. J Bone Joint Surg. 1985; 67:748-54.
-
Ramappa AJ, Lee FY, Tang P, Carlson JR, Gebhardt MC, Mankin HJ. Chondroblastoma of bone. J Bone Joint Surg Am. 2000; 82(8):1140-1145.

Abstracts for IOB Scientific Days, 27-29 August 2020
...
Un caz interesant de leucemie mieloidă cronică manifestată sub formă de criză blastică limfoidă B
Andreea Neculcea, Andreea Spînu, Diana Cisleanu, Anca Nicolescu, Cristina Enache, Ana Maria Vlădăreanu
Prezentăm cazul unui pacient de 46 de ani, care s-a prezentat la camera de gardă în decembrie 2019 pentru durere în cadranul st...
Metastaze pancreatice hipervasculare: sunt un diagnostic imagistic uşor?
Ioana G. Lupescu, Mirela Boroş, Florinela Ștefănescu
Obiectiv. De a revizui caracteristicile computer-tomografice (CT) şi de imagistică prin rezonanţă magnetică (IRM) ale metastazelor pancreatice hipervascularizate (MPH). De a discuta diagnosticul diferenţial al MPH. ...
Algorithm of diagnosis in bone metastasis of unknown primary origin – experience of a single clinical center over a five-year period
Chrysoula Antoniadou, Doina Mihaela Pop, Florinel Pop
În acest articol, prezentăm experienţa Secţiei de anatomie patologică a Spitalului Clinic de Ortopedie-Traumatologie şi TBC Osteoarticular Foişor, Bucureşti, în conduita de diagnostic al metastazelor ...
Aneurysmal cyst of bone, a complex histopathological lesion – case presentation, differential diagnosis and correlation of imaging data
Chrysoula Antoniadou, Doina Mihaela Pop, Florinel Pop
Chistul osos anevrismal (COA) este o leziune osoasă osteolitică, expansivă, uneori distructivă, dar autolimitantă, fiind constituită din spaţii chistice vasculare separate de o stromă fibroasă. Pr...