Boala polichistică renală autozomal-dominantă
Autosomal dominant polycystic kidney disease
Abstract
Autosomal dominant polycystic disease represents the most commonly encountered nephrologic pathology among genetic kidney disease. It is characterized by the emergence and uncontrolled growth of renal cysts due to a mutation occurring in one of the PKD1 and PKD2 genes. Often this nephrological pathology is accompanied by extrarenal manifestations, therefore these patients’ management requires a complex approach from nephrologist, cardiologist and general physician.Keywords
polycystic kidney diseaserenal cysttreatmentRezumat
Boala polichistică autozomal-dominantă reprezintă patologia nefrologică cel mai frecvent întâlnită dintre bolile genetice renale. Se caracterizează prin apariţia şi creşterea necontrolată a chisturilor renale din cauza unei mutaţii survenite în una dintre genele PKD1 şi PKD2. Deseori, această patologie nefrologică este însoţită de manifestări extrarenale, astfel încât managementul pacienţilor necesită o abordare complexă din partea nefrologului, cardiologului şi a medicului de familie.Cuvinte Cheie
boală polichistică renalăchisturi renaletratamentBoala polichistică renală autozomal-dominantă (ADPKD) este cea mai frecventă boală genetică dintre afecţiunile nefrologice. La nivel global afectează între 4 şi 7 milioane de pacienţi, fiind întâlnită în 7-15% dintre cazurile pacienţilor ce necesită hemodializă. Prevalenţa bolii în Europa este de 2,7:10.000, conform unei metaanalize recent publicate, afectând ambele sexe în egală măsură(1).
1. Etiopatogenie
ADPKD este o afecţiune caracterizată în primul rând de apariţia şi creşterea necontrolată a unor chisturi la nivel renal, bilateral, la care se asociază într-o rată variabilă diferite manifestări extrarenale.
Apariţia acestor chisturi are la bază mutaţii suferite la nivel genetic:
-
la nivelul genei PKD1, localizată pe braţul scurt al cromozomului 16, care codifică proteina policistina 1 (80-85% dintre cazuri);
-
la nivelul genei PKD2, localizată pe braţul lung al cromozomului 4, care codifică proteina policistina 2 (10-15% dintre cazuri);
la nivelul altor gene (GANAB, PMM2), importanţa acestora fiind în curs de cercetare(2-3).
Proteina policistina 1 şi policistina 2 formează complexul de policistine situat la nivelul cililor epiteliului tubular distal al nefronului (în principiu, la nivelul tubului colector), având rol în joncţiunea intercelulară şi în joncţiunea celulară – matrice extracelulară, respectiv în transportul transmembranar al electroliţilor (Ca2+).
Mutaţia care interesează una dintre cele două gene (PKD1 sau PKD2) produce disfuncţia acestui complex de policistine, având ca efect apariţia chisturilor prin următoarele mecanisme:
-
proliferare celulară necontrolată la nivelul epiteliului tubular şi apoptoza celulară;
-
modificarea polarităţii celulare;
-
transport transmembranar anormal de Ca2+ şi lichide, cu acumulare de lichid intracelular.
Degenerescenţa chistică apare în aproximativ 5% dintre nefroni, însă, odată cu creşterea chisturilor, acestea comprimă ţesutul interstiţial, rezultând ischemia şi fibroza la nivelul interstiţiului renal, iar în final, scăderea funcţiei renale(4).
2. Tabloul clinic
Deşi ADPKD este o boală multisistemică, deseori este asimptomatică (mai ales în stadiile incipiente) şi se depistează accidental în urma unor investigaţii imagistice (ecografie abdominală, CT abdominal), efectuate de rutină sau cu ocazia explorărilor efectuate pentru alte afecţiuni.
2.1 Manifestări renale
Caracteristica de bază a acestei afecţiuni este prezenţa chisturilor renale bilaterale.
Apariţia chisturilor este diferită, în funcţie de gena implicată. Astfel, la cei cu mutaţia PKD1, chisturile apar mai devreme (până la vârsta de 30 de ani), comparativ cu cei cu mutaţia PKD2 (până la vârsta de 50-60 de ani).
Creşterea chisturilor se face progresiv, în cazuri extreme, rinichii ajungând la dimensiuni impresionante (4.000 g faţă de greutatea normală de 120-140 g).
Prezenţa chisturilor, în special a celor voluminoase, poate genera multiple complicaţii:
-
durere lombară, în flancuri – de obicei cu intensitate mică, asociată sau nu cu masă abdominală palpabilă
-
hematurie macroscopică
-
40-50% dintre pacienţi prezintă cel puţin un episod;
-
o durată de 2-7 zile, care, de obicei, cedează spontan;
-
ruptură de chist
-
durere abdominală intensă, asociată cu hematurie macroscopică;
-
suprainfecţia chisturilor
-
semne de infecţie urinară înaltă (febră, durere lombară, stare generală alterată);
-
este dificil de diagnosticat (deseori urocultura este negativă), fiind necesare efectuarea de hemocultură şi/sau investigaţii imagistice pentru a confirma diagnosticul;
-
litiaza renală
-
este frecventă la pacienţii cu ADPKD (poate interesa până la 25-30% dintre pacienţi);
-
prin prezenţa acesteia, creşte numărul episoadelor de hematurie şi de durere colicativă;
-
diagnosticul necesită, deseori, explorare CT sau RMN deoarece este dificil de diferenţiat ecografic de chisturile cu pereţi calcificaţi;
-
scăderea funcţiei renale
-
alterarea funcţiei renale apare în decada a 4-a sau a 5-a de viaţă (în principiu, la cei cu mutaţia PKD1);
-
degradarea renală este mai rapidă decât în alte etiologii, fiind în corelaţie cu creşterea volumului renal;
-
progresia este spre boală renală cronică, în stadiul final fiind necesar tratament substitutiv renal (cel mai frecvent hemodializă)(5).
2.2. Manifestările extrarenale pot fi clasificate în două mari categorii: chisturi prezente la nivelul altor organe decât rinichii, respectiv modificări cardiovasculare (CV) şi cerebrovasculare.
În afară de rinichi, chisturile pot apărea şi în alte organe:
-
chisturile hepatice
-
sunt prezente în 75-80% din cazuri, iar de obicei nu cauzează disfuncţie hepatică;
-
apar mai târziu decât chisturile renale (la vârsta de peste 60 de ani);
-
sunt mai frecvent întâlnite la femeile multipare, ridicând suspiciunea de implicare hormonală în dezvoltarea acestora;
-
similar chisturilor renale, se pot suprainfecta; în asemenea cazuri, tratamentul antibiotic nu este suficient, necesitând drenaj chirurgical;
-
chisturi pancreatice, splenice (10%)
-
chisturi tiroidiene (10-15%)
-
chisturi la nivelul veziculelor seminale şi al prostatei (10%)
-
chisturi arahnoidiene (5-8%).
Spectrul bolilor cardiovasculare la pacienţii cu ADPKD este unul divers. Aceştia pot prezenta diferite valvulopatii (prolaps de valvă mitrală, insuficienţă mitrală), hipertrofie ventriculară stângă sau anevrisme vasculare (de aortă, coronare, iliace). Totuşi, afecţiunea cea mai importantă din această categorie este hipertensiunea arterială (HTA). HTA este prezentă la 75-80% dintre pacienţi în momentul depistării ADPKD, iar la restul apare odată cu instalarea bolii renale cronice. Mecanismul patogenic al HTA la pacienţii cu ADPKD este multifactorial. Un rol important îl are ischemia locală produsă de chisturile voluminoase, care duc la activarea sistemului renină-angiotensină-aldosteron, însă cercetările experimentale recente au identificat policistine şi la nivelul peretelui arterial, remodelarea vasculară în urma defectului genetic fiind un alt factor major în dezvoltarea HTA(5-6).
Manifestările cerebrovasculare, în special anevrismele cerebrale, sunt prezente în 20-30% dintre cazuri şi reprezintă cele mai de temut boli asociate, deoarece ruptura acestora se asociază cu o mortalitate de peste 50%. Anevrismele cerebrale sunt de 5 ori mai frecvente la pacienţii cu ADPKD decât la populaţia generală şi prezintă agregare familială.
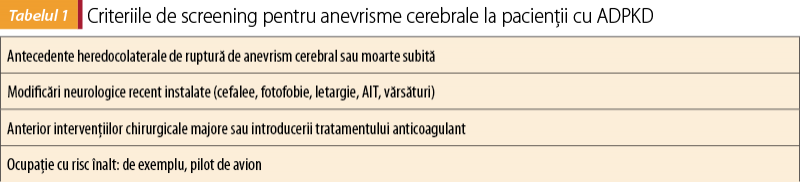
Odată cu apariţia posibilităţii folosirii metodelor imagistice pe scară largă, se recomandă un screening riguros al anevrismelor cerebrale ale pacienţilor cu risc(7). Criteriile de screening sunt redate în tabelul 1.
3. Diagnostic
Deşi ADPKD este o boală al cărei substrat genetic a fost elucidat, diagnosticul molecular nu este de rutină din cauza costurilor ridicate şi a heterogenităţii genetice. Se recomandă efectuarea testărilor genetice în cazul pacienţilor donatori de rinichi asimptomatici, dar cu istoric familial de boală polichistică renală.
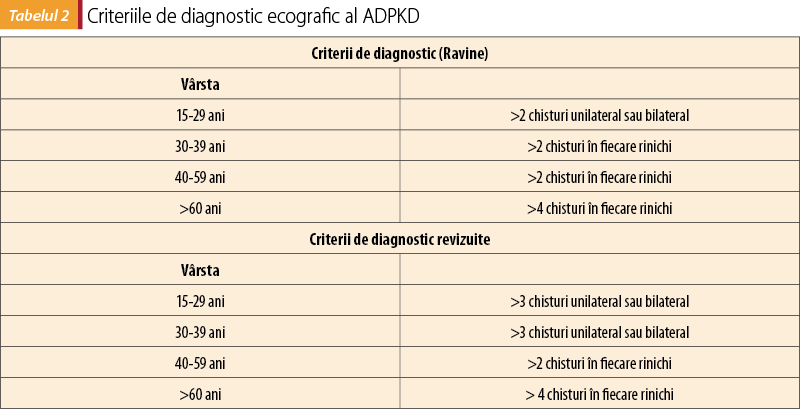
Stabilirea diagnosticului de ADPKD la un pacient cu o simptomatologie renală sugestivă şi/sau manifestări extrarenale cu istoric familial pozitiv are la bază modificările decelate prin ecografie renală. Criteriile de diagnostic ultrasonografic al ADPKD, ajustate în funcţie de vârstă, au suferit anumite modificări pe parcursul anilor şi sunt prezentate în tabelul 2(8).
4. Evoluţie
ADPKD este o boală cu evoluţie progresivă spre boala renală cronică în stadiu final. Odată cu creşterea chisturilor, apare şi o degradare a funcţiei renale, scăderea eGFR-ului corelându-se cu creşterea volumului renal. Deşi au fost identificaţi mai mulţi factori prognostici negativi (sexul masculin, diagnostic la vârsta sub 35 de ani, genotipul PKD1, prezenţa HTA la debut, chisturi suprainfectate, episoade de hematurie macroscopică), care contribuie la o creştere rapidă a volumului renal şi implicit la dezvoltarea şi agravarea bolii renale cronice, doar în ultimii 5 ani s-au dezvoltat diferite scoruri care stratifică pacienţii în diferite grupuri de risc(9).
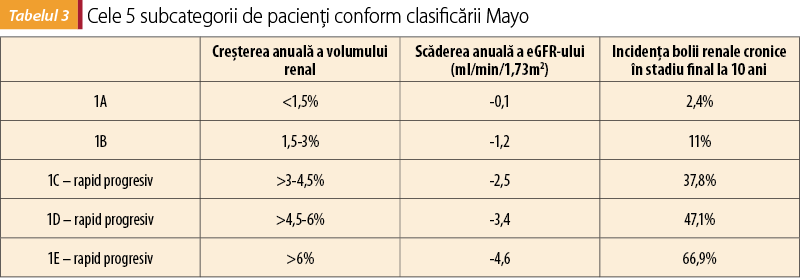
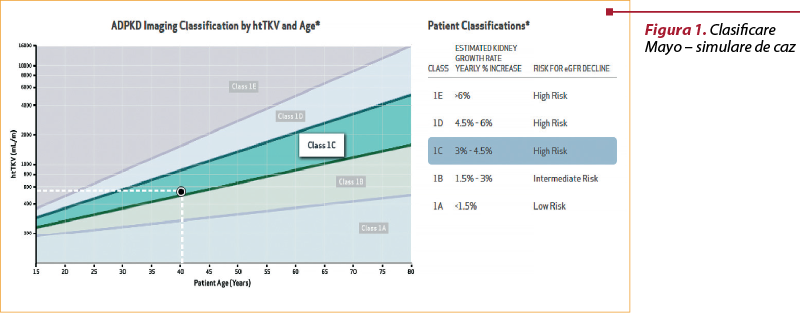
A. Clasificarea Mayo se bazează pe determinarea volumului renal prin metode imagistice de mare performanţă (de preferat, RMN). În funcţie de vârsta pacientului, respectiv volumul renal ajustat la înălţime, se descriu 5 subcategorii (1A-1E), prezentate în tabelul 3(10).
Folosirea acestui sistem de clasificare este foarte uşoară, fiind disponibile mai multe calculatoare online pentru stratificarea bolii. De exemplu, un pacient de 40 de ani, cu înălţimea de 1,70 m şi cu un volum renal de 1000 ml, se încadrează în subcategoria 1C conform unei simulări efectuate cu calculatorul online disponibil pe site-ul www.adpkdsim.org (figura 1).
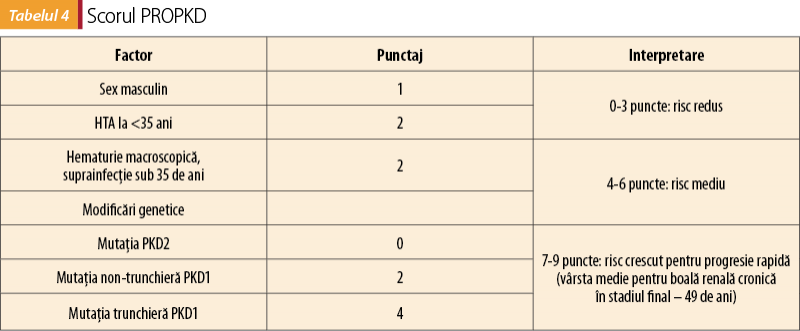
B. Scorul PROPKD reprezintă un alt sistem de stratificare a pacienţilor în funcţie de grupuri de risc. Această clasificare nu necesită investigaţii imagistice, însă se bazează pe determinări genetice. Factorii incluşi în determinarea scorului PROPKD sunt redaţi în tabelul 4(11).
C. Lungimea renală >16,5 cm, măsurată ultrasonografic la pacienţii sub 45 de ani, s-a dovedit a fi un factor predictiv pentru o evoluţie rapid progresivă. Deşi este cea mai uşoară metodă de aplicat în practica medicală, are acurateţea cea mai mică, fiind dependentă de evaluator, de experienţa acestuia(12).
5. Tratament
Managementul pacientului cu ADPKD necesită o abordare complexă.
Tratamentul conservator cuprinde recomandări igieno-dietetice, astfel:
-
regim hiposodat, hipocaloric, cu obţinerea/menţinerea unui indice de masă corporală de 25;
-
aport proteic de 0,8-1g/kgc/zi;
-
evitarea traumatismelor zonei lombare, în special în cazurile cu chisturi mari.
Tratamentul complicaţiilor este diferit, în funcţie de manifestările clinice:
-
hematuria macroscopicănu necesită, de obicei, tratament specific, este autolimitată şi cedează în 2-5 zile, rareori producând anemie;
-
suprainfecţia chisturilor necesită tratament antibiotic prompt, de preferat un antibiotic lipofil (fluorochinolone, trimetoprim);
-
tratamentul durerii rămâne un element-cheie în managementul acestor pacienţi, ameliorarea acesteia determinând o îmbunătăţire a calităţii vieţii. Tratamentul medicamentos (antialgic, AINS) este controversat, fiind necesară administrarea unei medicaţii potenţial nefrotoxice pe termen lung la un pacient cu funcţie renală fragilă. Tratamentul chirurgical/urologic, fie prin intervenţii minim-invazive (abordarea percutană ecoghidată a chisturilor cu evacuare şi sclerozare, decorticarea laparoscopică a chisturilor), fie prin nefrectomie uni- sau bilaterală, reprezintă o opţiune terapeutică în cazul chisturilor voluminoase;
-
terapia antihipertensivă este un alt element important, 75-80% dintre pacienţi necesitând administrarea de antihipertensive. Având în vedere implicarea sistemului renină-angiotensină-aldosteron în dezvoltarea HTA la pacienţii cu ADPKD, se recomandă administrarea de IEC sau sartani în prima linie, însă combinaţia celor două clase nu aduce beneficii suplimentare. Deşi dovezile nu sunt ferme, se recomandă evitarea utilizării de blocante de calciu şi/sau diuretice. Au fost demarate puţine studii randomizate privind valorile-ţintă ale tensiunii arteriale la aceşti pacienţi, Totuşi, pe baza rezultatelor existente, societăţile de specialitate din SUA recomandă controlul HTA în funcţie de diferiţi factori:
Control intensiv al TA≤110/75 mmHg la pacienţii
• cu vârsta de 18-50 de ani
• eGFR >60 ml/min/1,73 m2
• cu anevrism cerebral, afectare cardiovasculară
• clasa de risc Mayo 1C-E (rapid progresiv)
Control standard al TA≤ 130/85 mmHg(13-14).
Tratamentul patogenic al ADPKD cuprinde medicaţia care vizează încetinirea creşterii volumului renal prin inhibarea diferitelor căi de semnalizare intracelulare. În ultimii 10 ani au fost publicate numeroase cercetări cu diferite substanţe active: inhibitorii mTOR (rapamicină, everolimus), analogi de somatostatină cu rezultate parţial favorabile(15-16).
Un impact semnificativ privind tratamentul patogenic îl prezintă utilizarea antagonistului receptorului de tip 2 al vasopresinei (tolvaptan). Două mari studii randomizate, controlate, dublu-orb, cu două braţe (placebo versus tolvaptan), au inclus numeroşi pacienţi (1.445 de pacienţi – studiul TEMPO 3:4; 1.370 de pacienţi – studiul REPRISE) cu risc de progresie rapidă, conform criteriilor Mayo, fiind monitorizaţi timp de trei, respectiv un an. Rezultatele au fost net în favoarea braţului intervenţional: pacienţii care au beneficiat de administrare de tolvaptan au prezentat o rată de creştere a volumului renal statistic semnificativ mai mică decât braţul placebo(17-18). Astfel, în 2015, atât FDA, cât şi EMEA a aprobat utilizarea tolvaptanului (Jinarc®) la pacienţii cu ADPKD, cu vârsta sub 50 de ani, cu boală rapid progresivă, cu eGFR ≥30 ml/min/1,73m2, cu titrarea dozelor conform următoarei scheme:
-
Săptămâna 1: 45 mg à jeun + 15 mg seara
-
Săptămâna 2: 60 mg à jeun + 30 mg seara
-
Dacă tolerează à la longue: 90 mg à jeun + 30 mg seara.
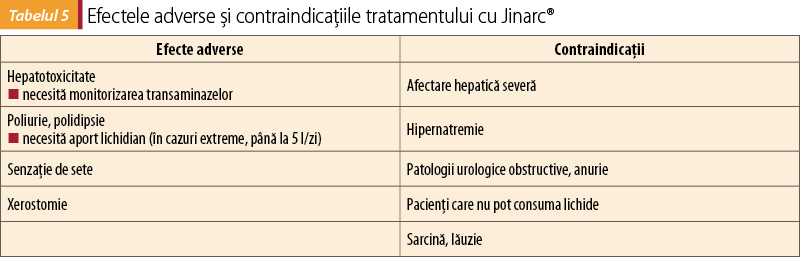
Efectele adverse şi contraindicaţiile sunt redate în tabelul 5.
Conflict of interests: The author declares no conflict of interests.
Bibliografie
- Solazzo A, Testa F, Giovanella S, Busutti M, Furci L, Carrera P, Ferrari M, Ligabue G, Mori G, Leonelli M, Cappelli G, Magistroni R. The prevalence of autosomal dominant polycystic kidney disease (ADPKD): A meta-analysis of European literature and prevalence evaluation in the Italian province of Modena suggest that ADPKD is a rare and underdiagnosed condition. PloS One. 2018; 13(1), e0190430.
- Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet. 2016;98(6):1193-207.
- Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H. Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol. 2017; doi:10.1681/ASN.2016121312.
- Halvorson CR, Bremmer MS, Jacobs SC. Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment. Int J Nephrol Renovasc Dis. 2010;3:69-83.
- Mititiuc I, Dominte A, Gorduza V, Segal L. Bolile genetice renale. In Adrian Covic ed. Nefrologie. principii teoretice şi practice, Casa editoriala Demiurg, Iaşi, 2011, 453-466.
- Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant. 2014;29(2):247-54.
- Flahault A, Joly D. Screening for Intracranial Aneurysms in Patients with Autosomal Dominant Polycystic Kidney Disease. CJASN. August 2019; 14 (8) 1242-1244.
- Gradzik M, Niemczyk M, Gołębiowski M, Pączek L. Diagnostic Imaging of Autosomal Dominant Polycystic Kidney Disease. Pol J Radiol. 2016 Sep 17;81:441-453.
- Grantham JJ, Torres VE, Chapman AB, Torres VE. Volume progression in polycystic kidney disease. N Engl J Med. 2006; 18;354(20):2122-2130.
- Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE; CRISP Investigators. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26:160-172.
- Cornec-Le Gall E, Frouget T, Vigneau C, Potier J, Jousset P, Guillodo MP, Siohan P, Terki N, Sawadogo T, Legrand D, Menoyo-Calonge V, Benarbia S, Besnier D, Longuet H, Férec C, Le Meur Y. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27(3):942-951.
- Bhutani H, Smith V, Rahbari-Oskoui F, Mittal A, Grantham JJ, Torres VE, Mrug M, Bae KT, Wu Z, Ge Y, Landslittel D, Gibbs P, O’Neill WC, Chapman AB; CRISP Investigators A comparison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease. Kidney Int. 2015;88:146-151.
- Chebib FT, Torres VE. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. CJASN. Nov. 2018; 13 (11) 1765-1776.
- Helal I. Treatment and Management of Autosomal Dominant Polycystic Kidney Disease. In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015 Nov. Chapter 3. Available from: https://www.ncbi.nlm.nih.gov/books/NBK373382/doi: 10.15586/codon.pkd.2015.ch3.
- Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2010; 363:830-840.
- Griffiths J, Mills MT, Ong AC. Long-acting somatostatin analogue treatments in autosomal dominant polycystic kidney disease and polycystic liver disease: a systematic review and meta-analysis, BMJ Open. 2020 Jan 9;10(1):e032620.
- Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, Perrone RD, Ouyang J, Blais JD, Czerwiec FS; TEMPO 3:4 Trial Investigators.Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD Stage: Results from the TEMPO 3:4 Trial. Clin J Am Soc Nephrol. 2016 May 6;11(5):803-11.
- Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, Ouyang J, McQuade RD, Blais JD, Czerwiec FS, Sergeyeva O; REPRISE Trial Investigators. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2017 Nov 16;377(20):1930-1942.

Actualitatea medicală
Mihai Mara
În scopul prevenirii îmbolnăvirilor şi recuperării capacităţii de muncă, asiguraţii pot beneficia de concediu şi indemnizaţie pentru carantină, conform reglementărilor cuprinse în Ordonanţa de Urgenţă a Guvernului nr. 158/2005 privind concediile şi indemnizaţiile de asigurări sociale de sănătate, cu modificăr...
Factori de risc implicaţi în apariţia nefropatiei induse de materialele de contrast
Andreea Varga, Dorina Nastasia Petra, Dragoş-Gabriel Iancu, Ioan Ţilea
Explorările invazive cardiovasculare reprezintă un grup de proceduri care au evoluat de la primul cateterism venos (Forssman, 1929) până la ultrasonografia intravasculară (IVUS) şi tomografia optică de coerenţă intraco...
Osteodistrofia renală – update
Claudia Floriana Suciu, Andreea Varga, Ioan Ţilea
Osteodistrofia renală se dezvoltă ca o consecinţă a dezechilibrării metabolismului fosfo-calcic la pacienţii cu boală renală cronică avansată. Aspecte ale patomecanismului osteodistrofiei renale sunt în continuare incomplet elucidate, însă descoperirile recente legate de importanţa şi acţiunea factorului d...
Factori de risc implicaţi în apariţia nefropatiei induse de materialele de contrast
Andreea Varga, Dorina Nastasia Petra, Dragoş-Gabriel Iancu, Ioan Ţilea
Explorările invazive cardiovasculare reprezintă un grup de proceduri care au evoluat de la primul cateterism venos (Forssman, 1929) până la ultrasonografia intravasculară (IVUS) şi tomografia optică de coerenţă intraco...
Osteodistrofia renală – update
Claudia Floriana Suciu, Andreea Varga, Ioan Ţilea
Osteodistrofia renală se dezvoltă ca o consecinţă a dezechilibrării metabolismului fosfo-calcic la pacienţii cu boală renală cronică avansată. Aspecte ale patomecanismului osteodistrofiei renale sunt în continuare incomplet elucidate, însă descoperirile recente legate de importanţa şi acţiunea factorului d...