Debut cu tromboze extensive în trombocitemia esenţială triplu negativă – dificultăţi de diagnostic. Prezentare de caz
Recurrent thrombosis onset in triple-negative essential thrombocythemia – more than a difficult diagnosis. Case report
Abstract
Essential thrombocythemia (ET) is a chronic myeloid neoplasm characterized by persistent nonreactive thrombocytosis (platelets ≥ 450x103/μL), specific changes in the bone marrow and driver mutations. The lack of mutations of JAK2, CALR and MPL driver genes is referred to as triple-negative genotype. The diagnosis is more difficult in triple-negative ET, requiring the exclusion of reactive thrombocytosis or further investigations to demonstrate clonality. The onset with recurrent thrombotic complications in this case led to an initial suspicion of chronic myeloproliferative neoplasm, but they raised issues regarding the differential diagnosis, leading to an extensive diagnosis workup.Keywords
essential thrombocythemiatriple negativemyeloid neoplasmRezumat
Trombocitemia esenţială (TE) este un neoplasm mieloid cronic caracterizat prin trombocitoză persistentă nonreactivă (trombocite ≥450x103/μL), modificări specifice ale măduvei osoase şi mutaţii driver. Lipsa mutaţiilor genelor driver JAK2, CALR şi MPL este denumită genotip triplu negativ. Diagnosticul este mai dificil în TE triplu negativă, fiind necesară excluderea trombocitozei reactive sau investigaţii suplimentare pentru a demonstra clonalitatea. Complicaţiile trombotice extensive în cazul prezentat au ridicat suspiciunea iniţială de neoplasm mieloproliferativ cronic, însă au pus probleme de diagnostic diferenţial, necesitând o abordare complexă şi multidisciplinară.Cuvinte Cheie
trombocitemie esenţialătriplu negativneoplasm mieloidIntroduction
Triple-negative essential thrombocythemia (ET) accounts for approximately 10% of ET cases(1). Compared to triple-negative primary myelofibrosis, triple-negative ET is described as an indolent disease, with a low incidence of vascular events and with a lower risk of evolution in acute leukemia (<1% in 10 years)(1).
The lack of one major criteria in triple-negative ET obliges the clinician to exclude any causes of reactive thrombocytosis or to demonstrate the clonality using next-generation sequencing (NGS) techniques(1). Most frequent non-driver mutations in triple-negative ET are TET2 and KIT(1).
Case report
We report the case of a 67-year-old female who presented in the emergency department in another hospital, complaining of nausea, lumbar and left lower limb pain. The patient had a positive history of cervical cancer 13 years before admission, treated with chemotherapy, radiotherapy and hysterectomy.
The clinical exam showed grade 2 splenomegaly and a non-palpable pulse at the left popliteus artery. No enlarged lymph nodes or hepatomegaly were revealed by the clinical exam.
The laboratory investigations are presented in Table 1. The complete blood count (CBC) showed extreme thrombocytosis, anemia and leukocytosis with neutrophilia. The increased erythrocyte sedimentation rate (ESR) and fibrinogen suggested an inflammatory syndrome. No significant changes were found in liver, kidney function and electrolytes.
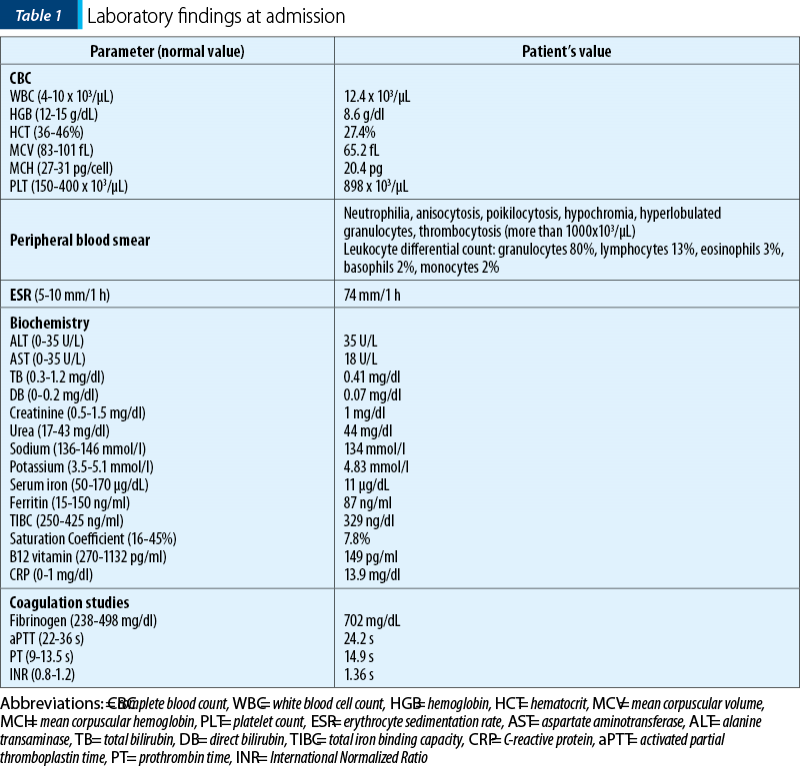
A computed tomography (CT) scan was performed, which revealed the presence of multiple thrombi located in both arterial and venous territories (abdominal aorta, left femoral artery, internal iliac artery, superior mesenteric artery, splenic vein) and splenic infarction (Figure 1). No tumors were detected.
Anticoagulation was promptly initiated with heparin given as an initial intravenous bolus of 5000 units, followed by a maintenance dose of 30,000-40,000 units per 24 hours by continuous intravenous infusion.
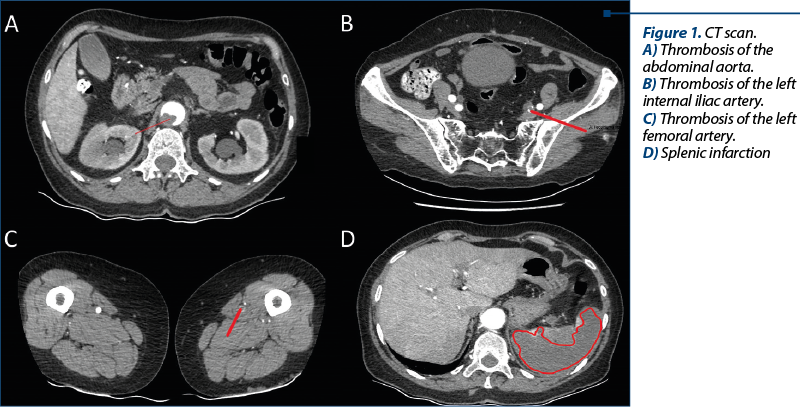
A radial approach angiography was performed. During the procedure, the patient developed hemiparesis and speech disorder. The head CT scan showed hypodensity in the superficial middle cerebral artery territory (Figure 2). There were no signs of intracerebral or subarachnoid hemorrhage, ventricles were in normal position, and cerebral-spinal fluid spaces looked normal.
At this moment, the patient was transferred to our center for mechanical revascularization. At admission to our hospital, the patient had global aphasia and right central facial palsy. Further investigations were performed to discover the etiology of the ischemic stroke.
Cervical-cerebral Doppler ultrasound did not reveal any atheromatous plaques. Cardiac echocardiography revealed no thrombi or vegetations. Holter monitoring revealed no episodes of flutter or atrial fibrillation. Therefore, the cardiovascular embolic mechanisms were excluded.
The CT scan previously performed showed no tumors. The tumoral markers (CA15-3, AFP, CEA and CA19-9) were normal, except for CA125, which was mildly elevated (48.3 U/ml). The breast ultrasonography and mammography did not reveal any malignant anomalies. The superior and inferior endoscopies were normal. Therefore, we excluded paraneoplastic syndrome or reactive thrombocytosis of malignant cause.
The screening for infectious disease and for thrombophilia was negative.
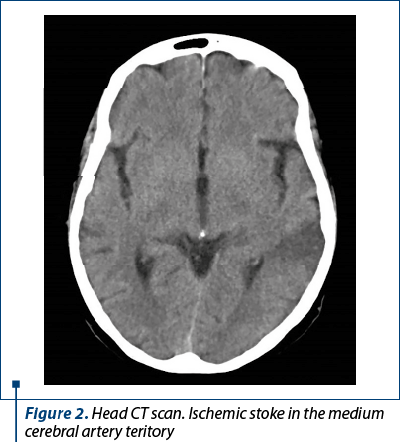
Therefore, we proceeded to investigate the patient for a hematological disorder. The peripheral blood smear showed marked thrombocytosis; no immature cells were spotted, differential blood count revealed neutrophilia and 2% basophils, 3% eosinophils (Figure 3). A bone marrow biopsy was performed, showing normal cellularity for her age, absent intrasinusoidal hematopoiesis, with a few large, prominent megakaryocytes with abundant mature cytoplasm and hyperlobulated nuclei. CD34 was positive in less than 1% of cells, and Gömöri trichrome stain showed no collagen or reticulin fibers (MF-0). Genetic mutations, including JAK2 V617F, MPL and CALR, were negative.\
The presence of recurrent thrombotic episodes, including splanchnic veins thrombosis, arterial cerebral stroke and splenomegaly, associated with persistent thrombocytosis and lack of any signs for reactive thrombocytosis, corroborated with bone marrow aspect and lack of criteria for other myeloproliferative neoplasms, pled for a diagnosis of essential thrombocythemia. Thus, the diagnosis of high-risk triple-negative ET was established, considering the patient’s age and her thrombotic history. Cytoreduction therapy with hydroxyurea was readily initiated to prevent recurrent thrombotic or hemorrhagic complications, in association with anticoagulant and antiplatelet agent that were previously recommended. The platelet count dropped steadily and the patient was discharged to be monitored in an outpatient care setting with recommendations to continue anticoagulants (acenocoumarin), aspirin and hydroxyurea.
At the follow-up, the patient developed hematological toxicity (mainly neutropenia and anemia). Anagrelide was introduced concomitantly at a starting dose of 0.5 mg and was to be further adjusted until a satisfactory balance between treatment toxicity and platelet count was obtained. Furthermore, acenocoumarin was switched to apixaban due to its higher predictability profile.
Discussion
The diagnosis of triple-negative essential thrombocythemia may be a challenge and requires the exclusion of reactive thrombocytosis or evidencing clonality(2). The NGS analysis has greatly improved the understanding of MPN pathogenesis and may offer the argument of molecular clonality.
Specific clinical data in corroboration with the hematological picture may offer the clues to diagnosis.
Triple-negative ET is an indolent disorder, with less frequent vascular events and with a low risk of leukemic evolution(1). In our case, the onset was sudden and severe, with extensive and recurring vascular events as the initial manifestation of the hematological disease.
The current guidelines recommend risk-adapted therapy. The risk factors in essential thrombocythemia are: history of thrombosis, advanced age (over 60 years old), the presence of JAK2 mutation, leukocytosis above 11x103/µL, and classical cardiovascular risk factors (smoking, arterial hypertension, diabetes mellitus)(3,4). Our patient had two risk factors for thrombosis, namely a history of thrombosis and advanced age. Therefore, she was classified as a high-risk patient. According to the current treatment algorithm, the standard treatment includes hydroxyurea, aspirin and anticoagulants(3).
After initiating the treatment, the patient’s evolution was favorable. The platelet count decreased and remained stable under treatment, and the thromboses were remitted.
Limitations
The study of clonality by NGS was not available.
Conclusions
We presented the case of a patient with triple-negative essential thrombocythemia who had both arterial and venous thromboses at the moment of diagnosis. After starting the treatment, the disease had a favorable evolution concordant to the reported data.
The case was presented at the 7th National Conference of Hemostasis and Thrombosis in Romania, in November 2021.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Cattaneo D, Croci GA, Bucelli C, et al. Triple-Negative Essential Thrombocythemia: Clinical Pathological and Molecular Features. A Single-Center Cohort Study. Front Oncol. 2021 Mar 12;11:637116.
-
Rokkam VR, Kotagiri R. Secondary Thrombocytosis. 2021 Aug 1. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 32809645. Available at: https://www.ncbi.nlm.nih.gov/books/NBK560810/
-
Corobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859.
-
Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia treatment algorithm. Blood Cancer. 2018;8:2.

Hipereozinofilia în limfoamele maligne non-hodgkiniene T-periferice – mai mult decât o modificare hematologică
Diana Emanuela Bonea, Cristina Mambet, Mihaela Găman, Iuliana Iordan, Andreea Neculcea, Alina Grigoroiu, Patricia Pîrvan, Dragoş-Claudiu Popescu, Onofrei Alexandru , Cristina Enache, Stejara Nicoleta Mihai, Ferea Roxana Cătălina , Alexandra Maria Baciu, Ana Maria Vlădăreanu
Eozinofilia este o modificare hematologică frecventă, asociată cu neoplaziile hematologice, cu tumorile solide sau cu boli autoim...
Aspecte IRM ale leziunilor PI-RADS 5 – experienţa unui centru terţiar
Sandra O. Jurcă, Gabriel Gluck, Ioana G. Lupescu
Cancerul de prostată este una dintre cele mai frecvente afecţiuni maligne în rândul bărbaţilor, pentru care se depun eforturi sporite, cu scopul de a îmbunătăţi rata de diagnosticare timpurie şi de a preveni cazu...
Riscul de malignitate în terapiile utilizate la pacienţii cu psoriazis
Ana Maria Alexandra Stănescu, Anca A. Simionescu, Marina Ruxandra Oţelea, Ioana Veronica Grăjdeanu
Psoriazisul este o boală inflamatorie cronică. Unii autori au sugerat că pacienţii cu psoriazis au un risc crescut de cancer. ...
Particularităţile diagnosticului în limfoamele primare ale sistemului nervos central (PCNSL)
Anca Nicolescu, Ana Maria Vlădăreanu, Horia Bumbea, Diana Cisleanu, Raluca Nistor, Ion Dumitru, Tiberiu Sultan
Limfomul primar al sistemului nervos central este o formă rară de limfom extranodal care poate afecta creierul, leptomeningele şi ochiul sau măduva spinării, fără a exista dovezi ale unei boli sist...
Current management of relapsed/refractory immune thrombocytopenia
Alina Mititelu, Minodora Onisâi, Anca Nicolescu, Ioachim Preda-Naumescu, Ana Maria Vlădăreanu
Trombocitopenia imună (TCI) este o afecţiune autoimună dobândită, cu potenţial hemoragic, caracterizată prin prezenţa autoanticorpilor antitrombocitari şi afectarea sintezei trombocitare. Adulţii prezintă adesea o ...