Eritroleucemia – cauză rară de leucemie acută
Minodora Onisâi, Cristina Enache, Andreea Neculcea, Horia Bumbea, Ana Maria Vlădăreanu
14 Decembrie 2019A 44-year-old man presented for repetitive epistaxis and marked physical asthenia. The clinical exam revealed only profuse sweating, moderately pale teguments and hepatosplenomegaly.
The CBC at admission showed severe pancytopenia: WBC 3800/mm³, hemoglobin 6.9 g/dl and platelet count 17,000/mm³. The biochemistry was unremarkable, with the exception of increased LDH (> 5x normal) and mild hepatocytolysis.
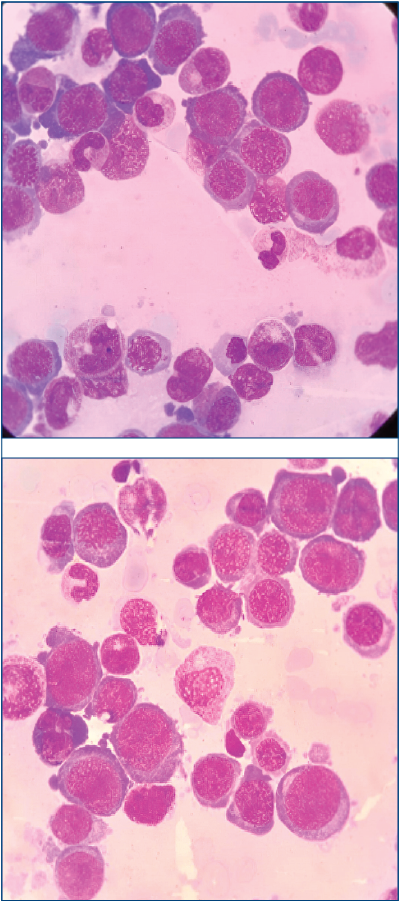
The peripheral blood smear revealed leukoerythroblastosis, with 13% blasts and 35 erythroblasts per 100 nucleated elements. The blast cells were medium-large in size, with basophilic cytoplasm, thin chromatin, nucleoli, some with granulation and rare with Auer bodies.
The bone marrow smear described hypercellular blood marrow, with 10% blast cells and markedly increased erythroblastic lineage (79%). The erythroblastic cells were large, with megaloblastic features, abundant basophilic cytoplasm with cytoplasmic projections and vacuolization, immature nuclear chromatin, and occasional nucleoli. The PAS staining was suggestive for AML6(1).
Immunophenotypic profile: glycophorin A positive, CD71 positive and CD33 positive, being suggestive for erythroleukemia – FAB classification(2) – AML6. This subtype is an exceptionally rare category of acute leukemia (3-8%), usually accompanied by unfavorable prognosis and evolution(1).
The cytogenetic exam revealed loss of the long arm of chromosome 5 – del (5q), associated with an adverse outcome(2).
The patient was successfully treated according to the “3+7” induction protocol(3) and then referred for consolidation with allogeneic bone marrow transplantation, performed from a matched unrelated donor, fortunately with a surprising favorable evolution.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Latif N, Salazar E, Khan R, Villas B, Rana F. The pure erythroleukemia: a case report and literature review. Clin Adv Hematol Oncol. 2010 Apr; 8(4):283-90.
- Schiffer C, Gurbuxani S. Classification of acute myeloid leukemia. UpToDate. 2019 Jul.
- Boddu P, Benton CB, Wang W, Borthakur G, Khoury JD, Pemmaraju N. Erythroleukemia – historical perspectives and recent advances in diagnosis and management. Blood Rev. 2018 Mar; 32(2):96-105.

Abstracts for “Stop Cancer”
The Third Translational Personalized Oncology Symposium – 10-12 May 2019, Bucharest...
Monitorizarea statusului oxidativ la pacienţii cu melanom uveal în timpul tratamentului oncologic
Maria Alexandra Budirinca, Maria Iuliana Gruia, Gabriela Murgoi, Rodica ANGHEL
Melanomul uveal este cea mai frecventă malignitate primară a ochiului la adulţi. Se cunosc puţine date despre etiologia sa. Factorii de risc includ predispoziţia organismului adult la diverse maladii,...
Screeningul naţional pentru cancerul de col uterin. Experienţă preliminară pentru screeningul cancerului de sân
Virgiliu Mihail Prunoiu, Oana Şaptefraţi, Rossana Iuliana Brătucu, Cristina Şaptefraţi, Eugen Bratucu, Alexandru Grigorescu, Maria Manuela Răvaş
Introducere. Cancerul de col uterin reprezintă o problemă de sănătate publică în România, anual fiind diagnosticate 4000 de femei ...
Hemorrhagic stroke in acute promyelocytic leukemia
Andreea Spînu, Iuliana Iordan, Minodora Onisâi, Mihaela Găman, Cristina Mambet, Diana Cisleanu, Ana Maria Neagu, Alina Mititelu, Andreea Neculcea, Cristina Enache, Lorena Pitiş, Raluca Nistor, Ana Maria Vlădăreanu
Leucemia acută promielocitară (LAP), o formă unică de leucemie acută promielocitară, este caracterizată de complicaţii hemoragice ...
Current management of relapsed/refractory immune thrombocytopenia
Alina Mititelu, Minodora Onisâi, Anca Nicolescu, Ioachim Preda-Naumescu, Ana Maria Vlădăreanu
Trombocitopenia imună (TCI) este o afecţiune autoimună dobândită, cu potenţial hemoragic, caracterizată prin prezenţa autoanticorpilor antitrombocitari şi afectarea sintezei trombocitare. Adulţii prezintă adesea o ...