Implicarea genomului papiloma virusului uman (hpv) în oncogeneza cancerului cervical
Involvement of Human Papillomavirus genome in oncogenesis of cervical cancer
Abstract
Genital HPV infection is the most common sexually transmitted viral infection among women, and persistent HPV infection with a high risk strain is the main etiology of malignant transformation of the cervix. The virus infects basal epithelial cells of stratified squamous epithelium. The integration of the viral genome (via E1/E2) in the host chromosomes is an important key-event in the HPV-induced carcinogenesis. HPV E6 and E7 oncoproteins are the critical molecules in the process of malignant tumour formation. Interacting with various cellular proteins, E6 and E7 influence fundamental cellular functions like cell cycle regulation, telomere maintenance, susceptibility to apoptosis, intercellular adhesion and regulation of immune responses. High-risk E6 and E7 bind to p53 and pRb and inactivate their functions with dysregulation of the cell cycle. Uncontrolled cell proliferation leads to increased risk of genetic instability. Usually, it takes decades for cancer to develop. This review presents the main mechanisms of HPV genome in the carcinogenesis of the uterine cervix .Keywords
cervical cancerHuman PapillomaviruscarcinogenesisRezumat
Infecția cu HPV este infecția virală transmisă cel mai frecvent pe cale sexuală la femei, iar infecția persistentă cu o tulpină cu risc ridicat este incriminată ca etiologie principală a cancerului de col uterin (cancerul cervical). Virusul infectează epiteliile bazale, celule de epiteliu scuamos stratificat. Integrarea genomului viral (via E1/E2) în cromozomii-gazdă este un eveniment-cheie în carcinogeneza indusă de HPV. Oncoproteinele HPV E6 și E7 sunt molecule critice în procesul de formare a tumorilor maligne. Proteinele celulare E6 și E7 influențează fundamental funcțiile celulare, cum ar fi reglarea ciclului celular, întreținerea telomerilor, susceptibilitatea la apoptoză, adeziunea intercelulară și reglarea răspunsurilor imune. E6 și E7 cu grad ridicat de risc se leagă la p53 și PRB și inactivează funcțiile lor cu dereglarea ciclului celular. Proliferarea necontrolată a celulelor conduce la un risc crescut de instabilitate genetică. De obicei, este nevoie de zeci de ani pentru a dezvolta un cancer. Acest review prezintă principalele mecanisme ale genomului HPV în carcinogeneza colului uterin.Cuvinte Cheie
cancerul colului uterinVirusul Papilomatos umancarcinogenezăIntroduction
Cervical cancer is the second most common cancer in women worldwide and the principal cancer of women in most developing countries, where 80 percent of cases occur mostly because of the inefficiency of screening programs. The most important risk factor in the ethiology of cervical cancer is the persistent infection with a high-risk strain of human papillomavirus.Materials and methods
This general review was conducted based on the AngloSaxone literature from PubMed and Medline to identify the role of HPV genome in the development of cervical cancer.Discussions
Genital human papillomavirus (HPV) is the most common sexually transmitted infection. Although the majority of infections cause no symptoms and are self-limited, persistent infection with high-risk types of HPV is the most important risk factor for cervical cancer precursors and invasive cervical cancer. The risk for persistence and progression to precancerous lesions varies by HPV type, with HPV 16 being more oncogenic than other high-risk HPV types. The presence of HPV in 99.7% of all cervical cancers implies the highest worldwide attributable fraction so far reported for a specific cause of any human cancer.HPV causes squamous cell cervical cancer (80% of all cervical cancer types) and its histologic precursor lesion are Cervical Intraepithelial Neoplasia [CIN] 1 (or low grade dysphasia) and CIN 2/3 (or moderate to high grade dysphasia) and cervical adenocarcinoma (and its precursor lesion, adenocarcinoma in situ [AIS]). HPV 16 and 18 are responsible for 70% of all cervical cancers. They are also responsible for others genital neoplasias like vaginal, vulvar, anal, and penian.
HPV is a non-enveloped, double-stranded DNA virus from the family of Papillomaviridae, with an 8 kb circular genome composed of six early ORFs (open reading frames) with role in viral transcription and replication (E1, E2, E4, E5, E6, E7), two late ORFs (L1,2-capsid proteins) and a non-coding long controlled region (LCR) that contains a variety of cis elements, which regulate viral replication and gene expression.
More than 100 HPV types have been identified, and about 40 can infect the genital tract. Based on their association with cervical cancer and precursor lesions, HPVs are grouped to high-risk (16, 18, 31, 33, 34, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, 73, 82) and low-risk HPV types (6, 11, 42, 43, 44, 54, 61, 70, 72, 81).
Natural history
Most genital HPV infections are benign, subclinical, and self-limited, and a high proportion of infections associated with low-grade cervical dysplasias also regress spontaneously(1). In 90% of cases, the immunologic system eradicate the infection in 1-2 years. By contrast, persistent cervical infection (infection detected more than once in an interval of 6 months or longer) with an oncogenic HPV type, especially HPV 16 and HPV 18, is the most important risk factor for progression to high-grade dysplasia, a precancerous lesion that should be treated to prevent the development of invasive cancer(2).
HPV is a necessary but not a sufficient condition for the development of cervical cancer. Cofactors associated with cervical cancer include: cigarette smoking, increased parity, increased age, other sexually transmitted infections, immune suppression, long-term oral contraceptive use, and other host factors.
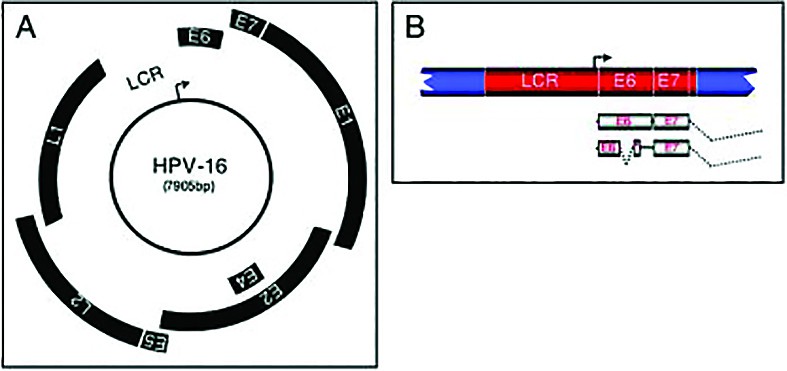
HPV integration into the host genome and Papillomavirus life cycle
To establish infection, the virus must infect basal epithelial cells of stratified squamous epithelium, that are long lived or have stem cell-like properties. Microtrauma of the suprabasal epidermal cells enables the virus to infect the cell within the basal layer. Once inside the host cell, HPV DNA replicates as the basal cells differentiate and progress to the surface of the epithelium. The viral genome maintains itself as an episome in basal cells, where the viral genes are poorly expressed. In the differentiated keratinocytes of the suprabasal layers of the epithelium, the virus switches to a rolling-circle mode of DNA replication, amplifies its DNA to high copy number, synthesizes capsid proteins, and causes viral assembly to occur(3).
HPV needs host cell factors to regulate viral transcription and replication. HPV replication begins with host cell factors which interact with the LCR region of the HPV genome and begin transcription of E6 and E7, two important oncogenes encoded by high risk-HPV. Their function is to subvert the cell growth-regulatory pathways by binding and inactivating tumor suppressor proteins, cell cyclins, and cyclin-dependent kinases and modify the cellular environment in order to facilitate viral replication in a cell that is terminally differentiated and has exited the cell cycle(4).
Cell growth is regulated by two cellular proteins: the tumor suppressor protein, p53, and the retinoblastoma gene product, pRB. Unlike in many other cancers, the p53 in cervical cancer is usually wild type and is not mutated. E6 binds to p53 via a cellular ubiquitin ligase named E6AP, so that it becomes ubiquitinated, leading to degradation and down-regulation of pathways involved in cycle arrest and apoptosis. This degradation has the same effect as an inactivating mutation. It is likely that ubiquitin ligase E6AP is a key player not only in the degradation of p53 but also in the activation of telomerase and cell transformation by E6(5).
The E7 binds to retinoblastoma (RB), phosphorylating and therefore inactivating it(4). Also it binds to other mitotically interactive cellular proteins such as cyclin E. Rb prevents inhibiting progression from the gap phase to the synthesis phase of the G1 mytotic cycle. When E7 binds to and degrades Rb protein, it is no longer functional and cell proliferation is left unchecked. The outcome is stimulation of cellular DNA synthesis and cell proliferation.
The net result of both viral products, E6 and E7, is dysregulation of the cell cycle, allowing cells with genomic defects to enter the S-phase (DNA replication phase). Further studies have documented additional evidence of the oncogenic potential of E6/E7, including the constant expression of E6/E7 and absence of corresponding regulatory mechanisms in tumour cell lines. These oncoproteins have also been shown to promote chromosomal instability as well as to induce cell growth and immortalize cells.
Next, the E5 gene product induces an increase in mitogen-activated protein kinase activity, thereby enhancing cellular responses to growth and differentiation factors. This results in continuous proliferation and delayed differentiation of the host cell.
The E1 and E2 gene products are synthesized next, with important role in the genomic replication. E1 has DNA helicase and ATPase activities. Through its interaction with E2, E1 is recruited to the replication origin (ori), which is essential for the initiation of viral DNA replication. E2 is a DNA binding protein which blocks transcription of the E6 and E7 genes. The loss of the repressive function of E2 is critical for the malignant progression because it leads to increased E6/E7 expression. The integration of the viral genome (via E1/E2) in the host chromosomes is an important key-event in the HPV induced carcinogenesis. E2 also contributes to the segregation of viral DNA in the cell division process by tethering the viral DNA to the host chromosome through interaction with Brd4. Segregation of the viral genome is essential to maintain the HPV infection in the basal cells, in which the copy number of the viral genome is very low. Then, a putative late promoter activates the capsid genes, L1 and L2(6).
Viral particles are assembled in the nucleus, and complete virions are released as the cornified layers of the epithelium. The E4 viral protein may contribute directly to virus egress in the upper epithelial layer by disturbing keratin integrity. In the replication process, viral DNA becomes established throughout the entire thickness of the epithelium but intact virions are found only in the upper layers of the tissue. This leads to acanthosis, parakeratosis, hyperkeratosis, and deepening of rete ridges, creating the typical papillomatous cytoarchitecture seen histologically.
Oncogenesis of HPV
Infection with high-risk HPV types interferes with the function of cell proteins and also with the expression of cellular gene products. Microarray analysis of cells infected with HPV-31 has shown that 178 cellular genes are up-regulated and 150 cellular genes are down-regulated by HPV(7).There are two main outcomes from the integration of viral DNA into the host genome that can eventually lead to tumour formation: blocking the cells apoptotic pathway and blocking synthesis regulatory proteins, leading to uncontrolled mitosis.
In benign lesions caused by HPV, viral DNA is located extrachromosomally in the nucleus. In high-grade intraepithelial neoplasias and cancers, HPV DNA is generally integrated into the host genome. Integration of HPV DNA disrupts or deletes the E2 ORF, which results in loss of its expression(8) and an increased expression of E6 and E7. In high-risk HPV types, the E6 and E7 proteins have a high affinity for p53 and pRB. Binding disrupts their functions, and alter cell cycle regulatory pathways, leading to cellular transformation. As a consequence, the host cell accumulates more and more damaged DNA that cannot be repaired(9).
The essential condition for the virus to determine a malign transformation is to persist in the tissue. In the outer layers of the epithelium, viral DNA is packaged into capsids and progeny virions are released to re-initiate infection. Because the highly immunogenic virions are synthesized at the upper layers of stratified squamous epithelia they undergo only relatively limited surveillance by cells of the immune system. In addition, E6 and E7 inactivate interferon (IFN) regulatory factor (IRF)(10), so that HPV viruses can remain as persistent.
Further studies have documented additional evidence of the oncogenic potential of E6/E7, including the constant expression of E6/E7 and absence of corresponding regulatory mechanisms in tumour cell lines. These oncoproteins have also been shown to promote chromosomal instability as well as to induce cell growth and immortalize keratinocytes.
Recently, several PDZ-domain-containing proteins have been identified to be targets of E6 proteins, including mammalian homologs of DLG (DLG1/hDLG) and Scribble (Scrib/Vartul), MUPP1, MAGI-1, -2, and -3, GIPC, PATJ, PTPN3 and PSD95. E6-induced degradation of these proteins potentially causes loss of cell-cell contacts mediated by tight junctions and thus contributes to the loss of cell polarity seen in HPV-associated cervical cancers(11).
In addition to the effects of activated oncogenes and chromosome instability, potential mechanisms contributing to transformation include methylation of viral and cellular DNA, telomerase activation, and hormonal and immunogenetic factors. Progression to cancer generally takes place over a period of 10 to 20 years.
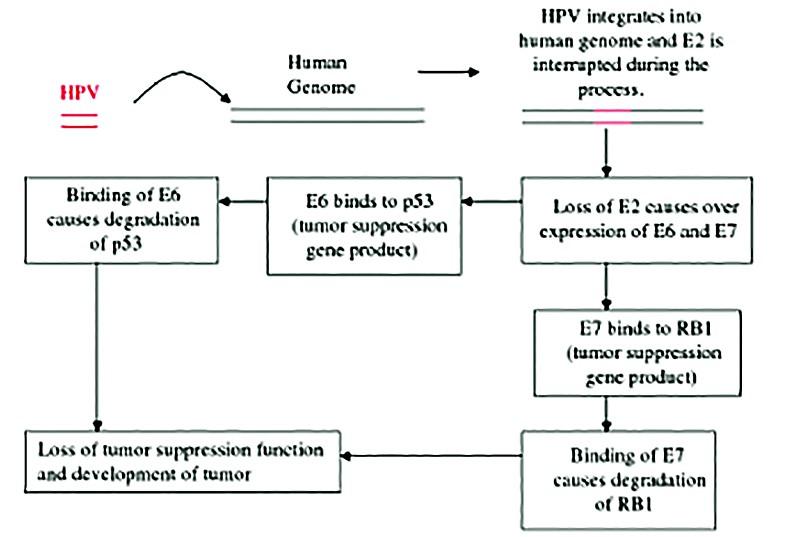
(“Mechanism of Oncogenesis. An Update on Tumorigenesis” - Domenico Coppola)
Conclusions
Cervical cancer is one of the best understood examples of how viral infection can lead to malignancy. Cervical carcinogenesis is a multifactorial process involving genetic, environmental, hormonal and immunological factors in addition to persistent HPV infection. Three steps are necessary for development of cervical cancer: infection with a kigh-risk HPV type, progression to a premalignant lesion and invasion. High-risk HPV-DNA integrate into the host genome and can lead to tumour formation by blocking the cells apoptotic pathway and blocking synthesis regulatory proteins leading to uncontrolled mitosis. HPV onco-proteins E6 and E7 are essential factors for HPV-induced cellular immortalization, transformation and carcinogenesis. Progression to cancer takes place over a very long period of time (decades), so the most important way to prevent its development is an efficient screening program of all women (regular Pap smears and gynecologic visits). n
Bibliografie
1. Baseman, J.G., and Koutsky, L.A. 2005). The epidemiology of human papillomavirus infections. J. Clin. Virol. 32(Suppl. 1): S16-S24.
2. Khan, M.J. et al. 2005. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J. Natl. Cancer Inst. 97:1072-1079.
3. Flores, E. R., B. L. Allen-Hoffman, D. Lee, C. A. Sattler, and P. F. Lambert. 1999. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 262:344-354.
4. Syrjänen, S. M., and K. J. Syrjänen. 1999. New concepts on the role of human papillomavirus in cell cycle regulation. Ann. Med. 31:175-187.
5. Thomas, M., D. Pim, and L. Banks. 1999. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 18:7690-7000.
6. McBride A. A., Oliveila J. G., McPhillips M. G. (2006). Partitioning viral genomes in mitosis: same idea, different targets. Cell Cycle 5, 1499–150210.4161/cc.5.14.3094.
7. Dietrich-Goetz W., Kennedy I. M., Levins B., Stanley M. A., Clements J. B. (1997). A cellular 65-kDa protein recognizes the negative regulatory element of human papillomavirus late mRNA. Proc. Natl. Acad. Sci. U.S.A. 94, 163–16810.1073/pnas.94.1.163.
8. Yoshinouchi, M., A. Hongo, K. Nakamura, J. Kodama, S. Itoh, H. Sakai, and T. Kudo. 1999. Analysis by multiplex PCR of the physical status of human papillomavirus type 16 DNA in cervical cancers. J. Clin. Microbiol. 37:3514-3517.
9. Halbert, C. L., G. W. Demers, and D. A. Galloway. 1991. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 65:473-478.
10. Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16, E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev 1998; 12: 2061–72.
11. Nakagawa S, Huibregtse JM. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol 2000; 20: 8244–53.

News from ASCO
Alexandru Grigorescu
An experimental monoclonal antibody has demonstrated promising results in neuroblastoma, according to new findings....
Tiolii albuminici - potențiali markeri ai statusului oxidativ în cancer
Maria Iuliana Gruia, Andreea Mirica
Stresul oxidativ a fost asociat, pentru o lungă perioadă de timp, cu fiziopatologia cancerului și a altor boli. Speciile reactive generate în exces duc la creșterea instabilității genomului care cauzează distrugeri ale A...
Radioterapia stereotactică - principii şi aspecte practice
Ciprian Enăchescu, Sena Yossi
Radioterapia stereotactică este o formă de iradiere externă care distribuie într-o şedinţă unică sau în câteva şedinţe doze mari de iradiere în volume-ţintă mici, tratament ce necesită o distribuţie a dozei extrem de precisă, astfel încât doza maximă să fie distribuită în volumul tumoral şi să scadă rapid per...
Tiolii albuminici - potențiali markeri ai statusului oxidativ în cancer
Maria Iuliana Gruia, Andreea Mirica
Stresul oxidativ a fost asociat, pentru o lungă perioadă de timp, cu fiziopatologia cancerului și a altor boli. Speciile reactive generate în exces duc la creșterea instabilității genomului care cauzează distrugeri ale A...
Radioterapia stereotactică - principii şi aspecte practice
Ciprian Enăchescu, Sena Yossi
Radioterapia stereotactică este o formă de iradiere externă care distribuie într-o şedinţă unică sau în câteva şedinţe doze mari de iradiere în volume-ţintă mici, tratament ce necesită o distribuţie a dozei extrem de precisă, astfel încât doza maximă să fie distribuită în volumul tumoral şi să scadă rapid per...