Aspecte clinice şi hematologice la pacienţii pediatrici diagnosticaţi cu anemie Fanconi – experienţa unui centru
Clinical and hematological aspects in pediatric patients diagnosed with Fanconi anemia – a single-center experience
Abstract
Inherited bone marrow failure syndromes (IBMFS) are rare hematological disorders that characteristically associate physical abnormalities, ineffective hematopoiesis and predisposition to cancer, accompanied by a range of congenital abnormalities. The most common are Fanconi anemia (FA), dyskeratosis congenita (DC), Diamond-Blackfan anemia (DBA) and Schwachman-Diamond syndrome (SDS). Most patients have specific clinical features, making them very important for establishing the correct diagnosis.Keywords
Fanconibone marrow failurethumb anomalieshematopoietic stem cell transplantationRezumat
Sindroamele de insuficienţă medulară congenitală reprezintă un grup de afecţiuni hematologice rare care se caracterizează prin insuficienţă medulară, având o predispoziţie pentru malignităţi. Cel mai des întâlnite sunt anemia Fanconi, diskeratoza congenitală, anemia Diamond-Blackfan şi sindromul Schwachman-Diamond. Majoritatea pacienţilor asociază şi malformaţii/anomalii scheletale, importante în stabilirea diagnosticului.Cuvinte Cheie
Fanconiinsuficienţă medularămalformaţii policetransplant de celule stemIntroduction
Inherited bone marrow failure syndromes (IBMFS) are considered pediatric diseases, though many patients are still diagnosed as adults. The most frequent are Fanconi anemia (FA), dyskeratosis congenita (DC), Schwachman-Diamond syndrome (SDS) and amegakaryocytic thrombocytopenia (which can often evolve to myelodysplastic syndrome or leukemia). Blackfan-Diamond anemia (DBA), severe congenital neutropenia (SCN) and thrombocytopenia with absent radii (TAR) are cytopenias involving only one line, rarely evolving to severe aplastic anemia, but with a high risk of developing acute leukemia(3,4).
The diagnosis of these diseases requires knowing the clinical signs, with specific laboratory evaluations and genetic testing. Patients with IBMFS have an increased risk of developing malignancies during their second lifetime period. The only treatment option for patients who have developed pancytopenia is hematopoietic stem cell transplantation(5,6).
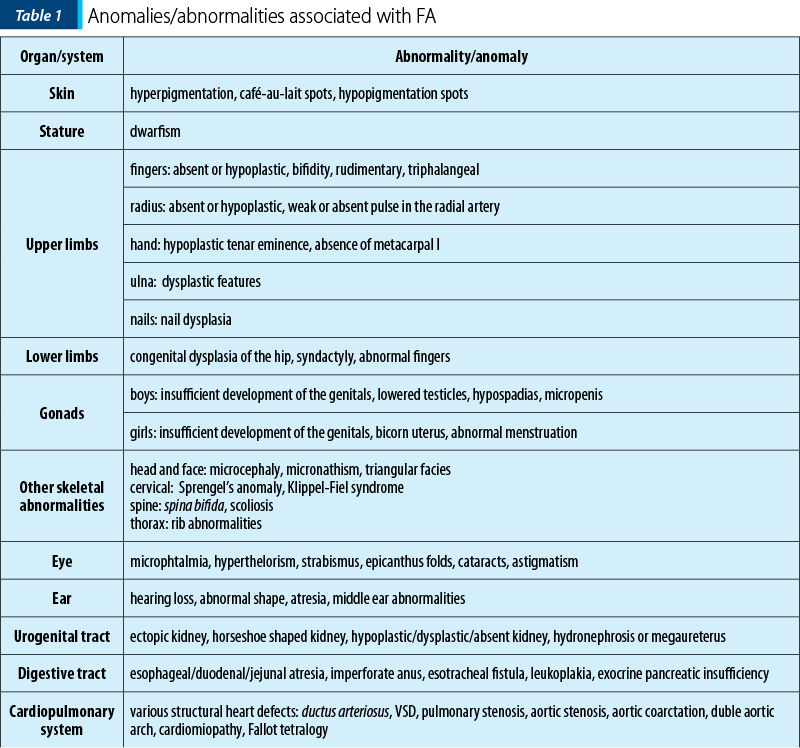
Fanconi anemia is a congenital disease with autosomal recessive inheritance. The most important characteristic is the predisposition to bone marrow failure and malignancies. The patients can have skeletal abnormalities, skin lesions, urinary tract malformations, intestinal or neurological symptoms, but approximately one-third of patients do not have somatic abnormalities. Bone marrow failure is most frequently diagnosed around the age of 7 years old, the patients presenting with cytopenia affecting one line or pancytopenia (anemia, thrombocytopenia, neutropenia). The clinical findings include café-au-lait spots on skin, skeletal abnormalities (short stature, microcephaly, hypoplastic thumbs, absence of thumbs/radius, polydactyly, syndactyly), other abnormalities (hypogenitalia, renal hypoplasia, microphtalmy, mental retardation) and chromosomal instability with ruptures, increasing the risk for acute leukemia or solid tumours(3,5,7).
The frequency of upper limbs abnormalities in FA patients is high, 50% of children having skeletal anomalies and 70% having upper limbs anomalies: thumbs deformities, radius malformation. Thumb malformation is graded in Table 2(1,2).

Objective
To evaluate the pediatric patients diagnosed with FA between 2002 and 2021 in the Pediatric Department of the Fundeni Clinical Institute, Bucharest, Romania.
Materials and method
We performed a retrospective study to analyze the clinical data, laboratory values, treatment options and the evolution of pediatric patients diagnosed with FA. The diagnosis was established on clinical findings and investigations: complete blood count, bone marrow biopsy, cytogenetic examination, a-feto-protein levels, imaging and genetic tests in complex cases. The conditioning regimens used in patients who received hematopoietic stem cell transplantation (HSCT) was adapted to each patient, according to the type of donor and procedure. Post-HSCT monitoring was perfomed using STR (short tandem repeats) technique to analyze chimerism. All parents signed the informed consent forms.
Results
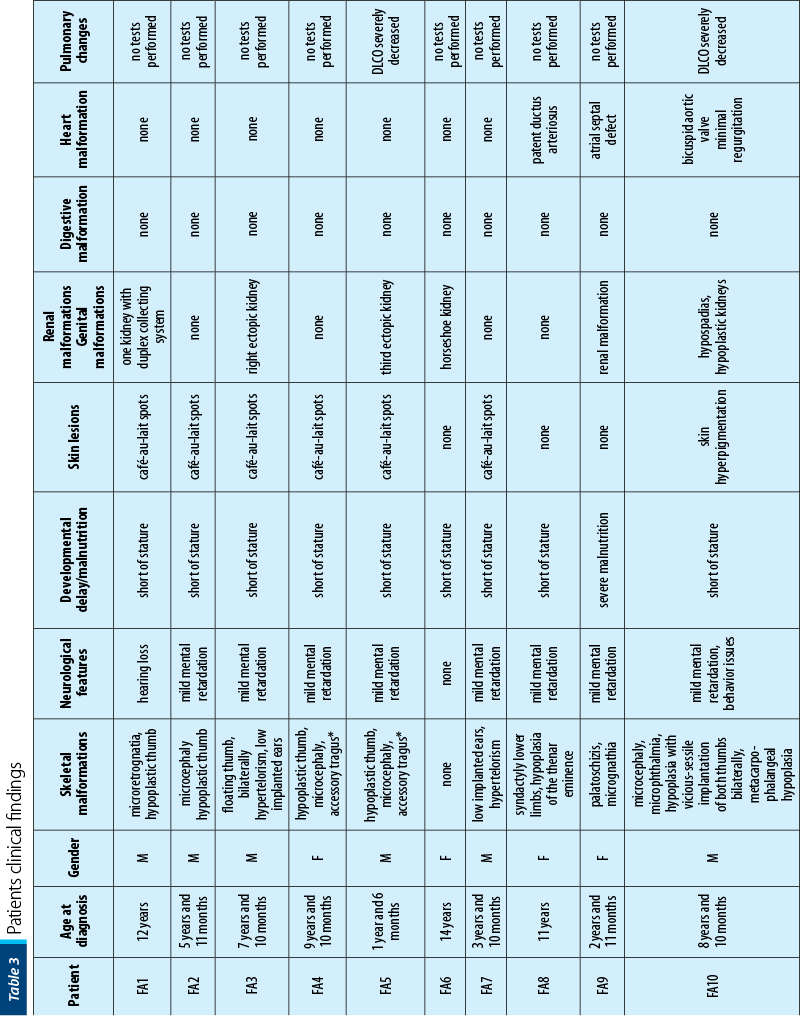
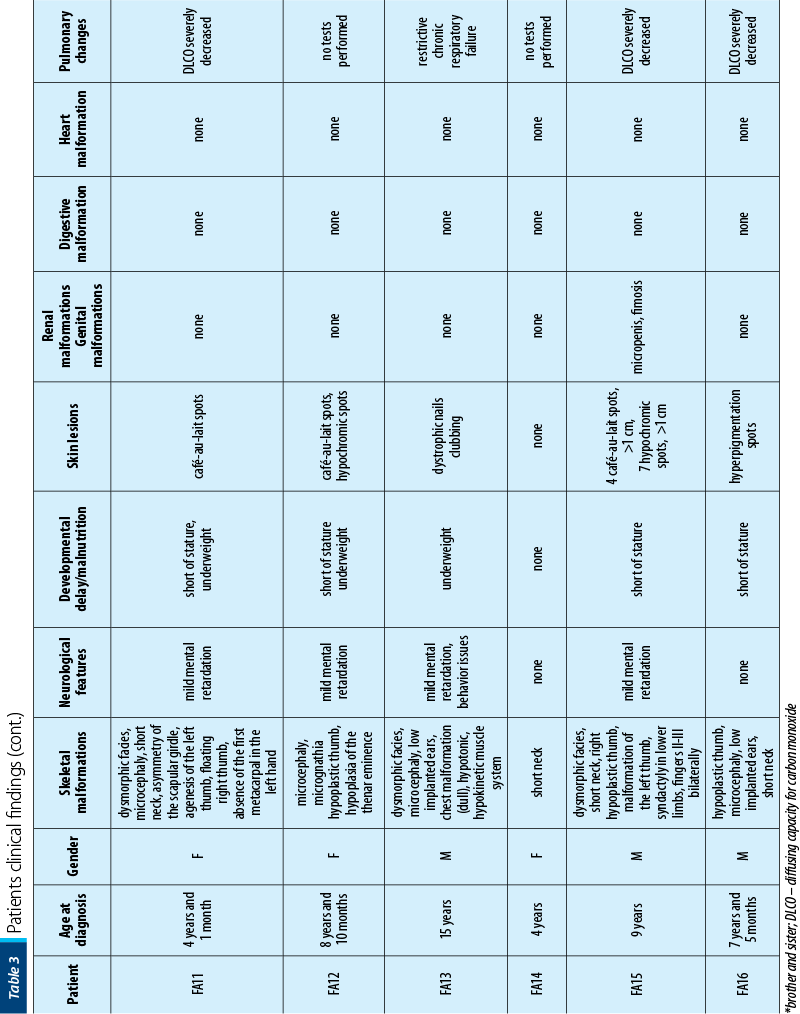
Between January 2002 and January 2021, 16 patients with AF (seven girls and nine boys), with a mean age at diagnosis of 7 years old (range: 1-15 years old) were diagnosed and treated in our department. Twelve out of 16 (75%) had bone malformations, nine out of 16 (56%) were short of stature, nine out of 16 (56%) had dysmorphic facies, six out of 16 (38%) had mental retardation, five out of 16 (31%) presented skin lesions, five out of 16 (31%) had renal malformations, three out of 16 (19%) had heart malformations and three out of 16 (19%) had genital tract abnormalities. Initially, we performed pulmonary evaluations only for patients who developed symptoms after HSCT, to exclude GvHD (graft versus host disease). Consequently, we extended the pulmonary screening to all IBMFS patients, several studies showing pulmonary changes as being characteristic in FA. In our study, six out of 16 patients had severely decreased DLCO (diffusing capacity for carbon monoxide), one of the patients having restrictive chronic respiratory failure due to thorax conformation.
The clinical findings are presented in Figures 1-6.
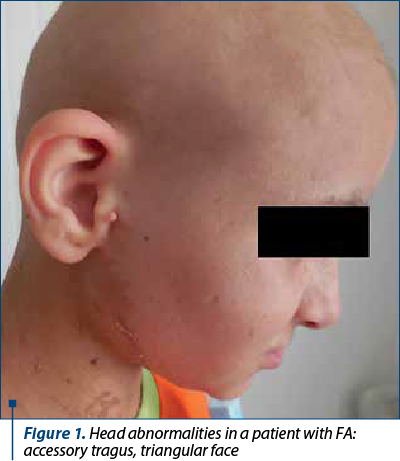
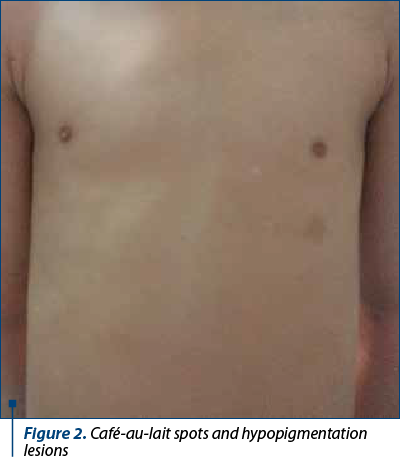
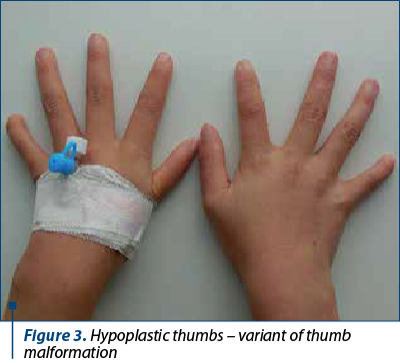
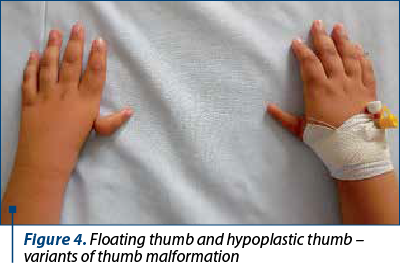

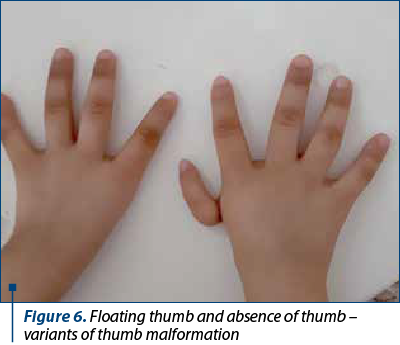
The patients’ data are presented in Table 3.
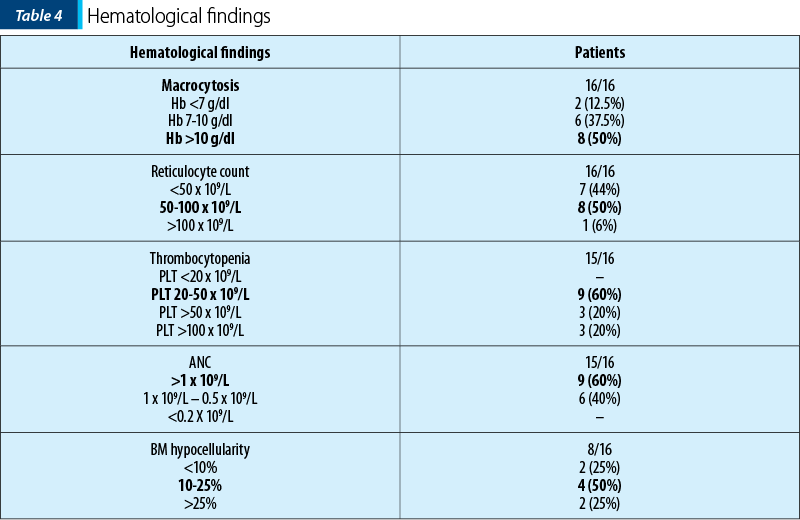
At onset, all patients had macrocytic anemia (median 10.8 g/dl, with median MCV of 95.65 fL), thrombocytopenia (median 41.5x109/L), neutropenia (median 1.4x109/L). The hematological findings are presented in Table 4. Bone marrow biopsy showed hipocellularity in all patients.
We performed HSCT procedure in 11 out of 16 patients, in four cases from a related donor and in seven cases from an unrelated donor. Four out of 16 patients did not present to further monitoring visits, one patient being currently monitored. Overall survival was 66.66%, with seven out 11 (63.6%) transplant recipients alive, with 100% donor chimerism at the last follow-up.
Discussion
Bone marrow failure syndromes – AF in this case – are complex conditions that require a multidisciplinary therapeutic approach.
Monitoring and correcting some of the physical abnormalities are necessary for improving the quality of life: orthopedic interventions for hand, finger, spine, lower limb abnormalities, plastic surgery interventions for certain facial abnormalities; monitoring and hearing aid in case of hearing abnormalities; monitoring and correction of cardiac, pulmonary, renal or gastrointestinal abnormalities.
Regularly asessments for establishing the indication for bone marrow transplantation are required. Also, monitoring and hormone replacement therapy for patients with various types of associated endocrine impairment are to be performed.
The long-term monitoring for the risk of cancer by including the patients in screening programs appropriate to individual risk should be performed.
It should be noted that bone marrow transplantation does not cure the disease, it only improves the quality of hematopoiesis, without reducing the risk of cancer; on the contrary, some studies suggest an increased risk of malignancy in these patients, secondary to chemotherapy used for the conditioning regimen.
Patients and their families require psychological counseling for adapting to the chronic condition, with evolutionary potential and involving many medical procedures.
Genetic counseling is needed to identify other family members affected by the same disease and the possibility of transmission to future generations(1,2).
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Tratat de Pediatrie (coord.: Prof. Dr. Doina Pleşca), Ed. 1, 2021.
-
Fanconi Anemia Guidelines for diagnosis and management, 4th Edition.
-
Dokal I, Vulliamy T. Inherited bone marrow failure syndromes. Haematologica. 2010;95(8):1236–1240. doi: 10.3324/haematol.2010.025619
-
Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101-122.
-
Bogliolo M, Surralles J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32-40.
-
Online Mendelian Inheritance in Man, OMIM (TM) [homepage on the Internet]. Baltimore e Bethesda: BeMcKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University and National Center for Biotechnology Information, National Library of Medicine. Available at: http://www.ncbi.nlm.nih.gov/omim/
-
Dufour C. How I manage patients with Fanconi anaemia. Br J Haematol. 2017;178:3247. https://doi.org/10.1111/bjh.14615

Sângerarea rectală la copii – prezentare de caz
Iulia Ţincu, Andrei Zamfirescu, Cristian Ioan Nedelcu, Anca Ioana Avram, Doina Pleşca
Polipii intestinali reprezintă mase tumorale proeminente în lumenul gastrointestinal, cu diverse aspecte histopatologice, de tip neoplazic sau non-neoplazic, epiteliale sau nonepiteliale. Forma clasică de prezentar...
Carcinomul corticosuprarenal – evoluţia fatală a unui tip rar de cancer la copil
Georgiana Scurtu, Magdalena Starcea, Mirabela Alecsa, Silvia Dumitraş, Antonela Ciobanu, Anca Ivanov, Adriana Mocanu, Ingrith Miron, Roxana Bogos
Carcinomul corticosuprarenal la copii este o tumoră rară, dezvoltată din celulele corticale suprarenale. Un pas foarte important...
Traumatismele abdominale la copii – experienţa unui singur centru pe o perioadă de un an
O. Bîcă, Diana Benchia, Klara Sârbu, Iulia Ciongradi, , S. Popa
Trauma este principala cauză a morbidităţii şi mortalităţii la copii. Datorită tehnologiei moderne, ghidurilor clinice şi intervenţiilor minim invazive, managementul traumei este diferit actualmente faţă de acum câţiva a...
Traumatismele abdominale la copii – experienţa unui singur centru pe o perioadă de un an
O. Bîcă, Diana Benchia, Klara Sârbu, Iulia Ciongradi, , S. Popa
Trauma este principala cauză a morbidităţii şi mortalităţii la copii. Datorită tehnologiei moderne, ghidurilor clinice şi intervenţiilor minim invazive, managementul traumei este diferit actualmente faţă de acum câţiva a...