Fetal congenital heart abnormalities (CHA) remain the most common congenital malformations encountered at birth and are the leading cause of infant mortality in developed countries. Conotruncal CHA comprises a broad category of congenital heart diseases, being present in a proportion of 10-12% of the total of postnatally diagnosed CHA. However, the prenatal diagnosis of these cardiac anomalies is more difficult to obtain because most of these abnormalities have a normal four-chamber appearance during routine fetal ultrasound screening. Early diagnosis and appropriate therapeutic behavior of severe and critical CHAs are essential to improve outcome.
MATERNAL-FETAL MEDICINE
Dificultăţi de diagnostic ecografic al malformaţiilor cardiace conotruncale fetale
Difficulties in ultrasound diagnosis of fetal congenital conotruncal heart malformations
First published: 23 iunie 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.2.2.2018.1810
Abstract
Rezumat
Anomaliile cardiace congenitale (ACC) fetale rămân cele mai frecvente malformaţii congenitale întâlnite la naştere şi reprezintă principala cauză a mortalităţii infantile în ţările dezvoltate. ACC conotruncale însumează o categorie largă de boli cardiace congenitale, fiind întâlnite în proporţie de 10-12% din totalul de ACC diagnosticate postnatal. Cu toate acestea, diagnosticul prenatal al acestor anomalii este mai dificil, deoarece majoritatea acestor anomalii au un aspect normal al imaginii de patru camere în timpul screeningului ecografic fetal de rutină. Diagnosticarea precoce şi conduita terapeutică adecvată a ACC grave şi critice sunt esenţiale pentru îmbunătăţirea rezultatului.
Introducere
Malformaţiile cardiace conotruncale sunt anomalii congenitale ale tractului de ejecţie cardiac, care includ defecte precum tetralogia Fallot (TF), atrezia pulmonară cu defect septal ventricular, ventriculul drept cu dublă cale de ejecţie (DORV), ventriculul stâng cu dublă cale de ejecţie (DOLV), trunchiul arterial comun, transpoziţia de vase mari (TGA), defectele septale ortopulmonare şi arcul aortic întrerupt de tip B(1).
Acest grup de defecte cardiace este mai frecvent întâlnit la feţii care asociază anomalii cromozomiale precum trisomiile 13, 16 şi 18, deleţii ale cromozomului 9 sau deleţia 22q 11.2 – sindromul DiGeorge. De asemenea, ACC conotruncale se asociază cu diverse malformaţii extracardiace, precum cele ale sistemului nervos central (holoprozencefalie, agenezie de vermis, microcefalie, chist de plex coroidian), ale tubului gastrointestinal şi defecte de perete abdominal (omfalocel, atrezie de esofag, atrezie duodenală, hernie diafragmatică, gastroschizis), malformaţii ale sistemului urogenital (rinchi polichistic, agenezie renală), ale sistemului osos (picior strâmb) sau cu alte anomalii, precum hidrotoraxul sau situs inversus(2).
Din punctul de vedere al dezvoltării embriologice, regiunea conotruncală reprezintă tractul de ejecţie al cordului aflat în dezvoltare şi cuprinde două subsegmente miocardice, conul cardiac şi trunchiul arterial. Conul cardiac reprezintă segmentul miocardic cuprins între ventricule şi valvele semilunare, aflat inferior de valvele aortice/pulmonare. Trunchiul arterial este un segment fibros, localizat superior de valve, aflat în continuitate cu aorta ventrală (sacul aortic), din care se vor dezvolta arterele mari ale inimii. Prima etapă embriologică în dezvoltarea regiunii conotruncale este septarea conului cardiac şi a trunchiului arterial, cu formarea a patru trunchiuri şi a două „perniţe” endocardiceconice. Perniţa conică endocardică stângă (localizată inferior), respectiv perniţa endocardică dreaptă (localizată superior) se vor uni şi vor forma septul conotruncal. Perniţele endocardice intercalate vor da naştere valvelor semilunare. În continuare, are loc o rotaţie a joncţiunii conoventriculare, respectiv a joncţiunii conotruncale, ambele în direcţia acelor de ceasornic, aproximativ la 110 grade. Rotaţia conoventriculară aduce aorta în continuare cu ventriculul stâng, respectiv artera pulmonară în continuare cu ventriculul drept. Rotaţia conotruncală stabileşte relaţia corectă dintre vasele mari ale inimii (aorta localizată la stânga şi posterior de artera pulmonară). O etapă importantă este şi absorbţia conului cardiac. Într-o inimă normală, conul subaortic este absorbit complet. În dezvoltarea regiunii conotruncale, celulele crestelor neurale joacă un rol important(3,4).
Referitor la dezvoltarea anomaliilor cardiace conotruncale, trunchiul arterial comun apare în urma eşecului diviziunii septului aortopulmonar. Septul infundibular care se dezvoltă din septul conal se dezvoltă anterior şi superior şi apar elemente de TF. În cazul DORV, rotaţia conoventriculară se bazează pe ce con persistă; dacă ambele conuri persistă, amândouă se vor naşte din ventriculul drept. În DOLV, ambele conuri sunt absente şi ambele artere sunt localizate posterior şi se dezvoltă din ventriculul stâng. În D-TGA, nu apare rotaţia conoventriculară, persistă conul subaortic, cu o resorbţie completă a conului subpulmonar. Defectele septale conoventriculare se caracterizează prin absenţa septului infundibular, cu apariţia defectelor septale ventriculare subinfundibulare, subpulmonare sau de malaliniament. Este important de ştiut faptul că asocierea ACC conotruncale cu anomaliile faciale sau ale timusului este frecvent întâlnită, întrucât timusul şi glandele paratiroide sunt, la rândul lor, dezvoltate din arcurile faringiene 3 şi 4, asemănător regiunii conotruncale a cordului(3,4).
Diagnosticul ultrasonografic al celor mai întâlnite ACC conotruncale
Ecocardiografia fetală (EF) are un rol în îmbunătăţirea rezultatelor chirurgicale la anumiţi feţi cu boli cardiace congenitale şi, prin urmare, contribuie la reducerea morbidităţii şi a mortalităţii neonatale. Anomaliile cardiace conotruncale sunt anomalii structurale care implică tractul de ejecţie al inimii, incluzând o gamă largă de fenotipuri, dintre care cele mai frecvente sunt tetralogia Fallot (TF) şi transpoziţia marilor artere (TGA). Majoritatea acestor anomalii pot pune viaţa în pericol imediat după naştere şi, de obicei, necesită intervenţie chirurgicală timpurie. Defecte cardiace congenitale cum ar fi dextro-TGA, TF, ventriculul drept cu dublă cale de ejecţie sau trunchiul arterial comun pot avea o imagine ecografică normală de patru camere. Aşadar, o examinare ecocardiografică în detaliu − şi în special examinarea conexiunii ventriculoarteriale − este de o importanţă capitală(6).
Diagnosticul ecografic al defectelor structurale cardiace fetale a beneficiat de o îmbunătăţire în ultima perioadă, datorită politicii de screening prenatal pentru malformaţii cardiace în timpul ultrasonografiei de rutină. Acesta este un factor foarte important care poate afecta incidenţa ACC, în cazurile în care se indică întreruperea sarcinii(6).
Progresul în tehnologia imagistică cu ultrasunete a îmbunătăţit semnificativ diagnosticul prenatal al malformaţiilor cardiace fetale. EF este o procedură costisitoare, de lungă durată, care necesită un operator de înaltă calificare şi cu experienţă. Astfel, nu este fezabil să se efectueze EF la toate femeile însărcinate ca o procedură de rutină în ţările în curs de dezvoltare. De aceea, este foarte important să se definească indicaţiile pentru efectuarea EF. Indicaţiile sunt factorii fetali, materni şi ereditari care reprezintă riscuri mari pentru a avea un făt cu ACC. Cu toate acestea, s-a raportat că majoritatea feţilor cu ACC se află în grupul cu risc scăzut şi au fost suspectaţi în timpul screeningului ultrasonografic de rutină efectuat în al doilea trimestru de sarcină(6).
D-TGA
D-TGA este a doua cea mai comună boală cardiacă congenitală cianotică diagnosticată în primul an de viaţă. Incidenţa raportată este de 315 la un milion de naşteri vii. D-TGA este observată predominant la sexul masculin, cu un raport între sexe de la 2:1 la 3:1. Embriologic, s-a presupus că eşecul rotaţiei conoventriculare şi persistenţa conusului subaortic, cu resorbţia completă a conusului subpulmonic, au ca rezultat această anomalie conotruncală. Spre deosebire de alte anomalii conotrunculare, d-TGA este rareori asociată cu anomalii cromozomiale sau malformaţii extracardiace. Diabetul matern este considerat a fi un factor de risc asociat cu o creştere de aproape trei ori a riscului pentru TGA(7).
Diagnosticul antenatal al acestei anomalii depinde în mod esenţial de demonstrarea tracturilor de ejecţie paralele sau a absenţei încrucişării marilor artere. Prin includerea următorilor paşi în evaluarea de rutină a inimii fetale, această anomalie poate fi diferenţiată în mod fiabil de TGA corectată fiziologic. Aceşti paşi sunt: a) evaluarea situsului şi a axei cordului; trebuie menţionat faptul că foramen ovale se deschide în atriul stâng, apare o herniere a septului interatrial dinspre dreapta spre stânga, cu apariţia unui atriu drept mai mare; b) demonstrarea concordanţei atrioventriculare, cu identificarea ventriculelor stâng (VS), respectiv drept (VD); ventriculul drept se recunoaşte după prezenţa bandei moderatoare, inserţia mai joasă (spre apex) a valvei tricuspide, comparativ cu valva mitrală, neregularităţile suprafeţei endocardice a VD, comparativ cu VS (acesta având o cavitate sub formă triunghiulară); c) demonstrarea modelului de ramificare a vaselor mari(7).
În concluzie, această anomalie cardiacă conotruncală poate fi diagnosticată prenatal facil, dacă există o abordare ultrasonografică sistematică, respectând nişte criterii de diagnostic.
Trunchiul arterial comun
Trunchiul arterial comun este un defect cardiac congenital caracterizat prin prezenţa unui singur trunchi arterial, o singură valvă arterială şi din care se nasc direct arterele coronare, sistemice şi una sau ambele artere pulmonare. Această anomalie conotruncală este rezultatul unei septări incomplete (persistentă) a porţiunii distale a tractului cardiac de ejecţie al inimii embrionare în aorta şi artera pulmonară. Trunchiul arterial comun este o anomalie cardiacă congenitală cianotică, caracterizată prin creşterea debitului sangvin pulmonar. Repararea chirurgicală precoce în perioada neonatală scade morbiditatea şi mortalitatea şi previne sechelele de lungă durată ale circulaţiei pulmonare şi insuficienţa cardiacă(8).
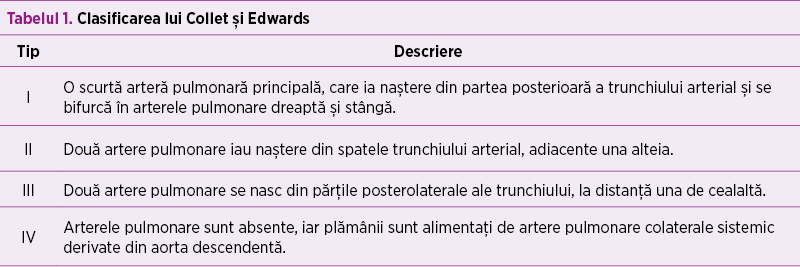
Collet şi Edwards au clasificat anomalia arterială truncală în funcţie de originea anatomică a arterelor pulmonare şi de relaţia spaţială dintre aceste vase (tabelul 1). În 1965, Van Praagha propus o altă clasificare anatomică, ce ia în considerare şi prezenţa sau absenţa unui defect de sept ventricular (DSV) în asociere cu trunchiul arterial comun.
Anomaliile cardiace conotruncale, cum ar fi trunchiul arterial comun, pot fi diagnosticate prenatal cu o precizie de până la 80% din cazurile în care se utilizează mai multe planuri sonografice, cartografierea fluxului color Doppler şi utilizarea Dopplerului pulsat. Experienţa în ecocardiografia fetală este o necesitate pentru evaluarea prenatală, deoarece imaginea cu patru camere, esenţială în screeningul prenatal cardiac, este normală în până la 40-50% din cazurile în care există un defect cardiac congenital major. Mai mult decât atât, în anomaliile cardiace conotruncale, imaginea de 4 camere este anormală într-o proporţie de doar 30%(8).
Diagnosticul ecocardiografic prenatal al trunchiului arterial comun a fost raportat încă din primul trimestru de sarcină (folosind ultrasonografia transvaginală de înaltă rezoluţie), la 16 săptămâni de gestaţie, la un făt cu translucenţa nucală crescută şi focare ecogene bilaterale şi la 18 săptămâni de gestaţie la un făt cu restricţie de creştere intrauterină. Cu toate acestea, majoritatea cazurilor sunt diagnosticate între 20 şi 25 de săptămâni de amenoree(8).
Vizualizarea unui vas arterial unic „călare” pe un DSV (mai degrabă decât o aortă distinctă şi artera pulmonară) în timpul examinării tracturilor de ejecţie este o constatare comună a trei defecte cardiace: trunchiul arterial comun, tetralogia Fallot şi atrezia pulmonară cu DSV. În tetralogia Fallot şi atrezia pulmonară cu DSV, o arteră pulmonară distinctă care iese din ventriculul drept este prezentă anatomic; cu toate acestea, gradul de stenoză/atrezie pulmonară poate fi de aşa natură, încât să nu poată fi recunoscut prin ultrasunete şi poate fi identificat doar un singur vas (aorta) care rezultă din inima fetală. Prin urmare, diagnosticul diferenţial pentru trunchiul arterial comun se bazează pe demonstrarea ultrasonografică a arterei pulmonare principale (sau cel puţin a uneia dintre ramurile sale) că se naşte din trunchiul arterial unic(8).
Trunchiul arterial comun este o anomalie conotruncală complexă cu mai multe subtipuri. Aşadar, diagnosticul prenatal necesită examinarea secvenţială a mai multor planuri de scanare şi un proces de reconstrucţie mintală a relaţiilor lor spaţiale, necesitând astfel expertiză şi cunoaştere în ecocardiografia fetală. Totuşi, diagnosticul poate fi facilitat de noile tehnici ultrasonografice 3D şi 4D, inclusiv afişarea multiplanară şi imagistica tomografică cu ultrasunete(8).
Tetralogia Fallot
Tetralogia Fallot (TF) este o malformaţie cardiacă congenitală care constă într-o comunicare interventriculară, cunoscută şi sub numele de defect septal ventricular, obstrucţia tractului de ejecţie al ventriculului drept, aorta „călare” pe septul interventricular şi hipertrofia ventriculară dreaptă. Apare la 3 din 10.000 de nou-născuţi vii şi reprezintă 7-10% dintre toate malformaţiile congenitale cardiace. Etiologia este multifactorială, însă ca factori de risc au fost raportate diabetul matern netratat, fenilcetonuria şi consumul de acid retinoic. Anomaliile cromozomiale asociate pot include trisomiile 21, 18 şi 13, dar experienţa recentă arată o asociere mult mai frecventă a microdeleţiilor cromozomului 22. Riscul reapariţiei în familie este de 3%(9).
Tetralogia Fallot poate fi diagnosticată antenatal încă din primele 12 săptămâni de gestaţie. Cu toate acestea, într-un studiu populaţional, s-a demonstrat că doar jumătate dintre cazuri au fost detectate în timpul screeningului ultrasonografic(9).
Descoperirea ultrasonografică a unei aorte mărite cu o formă de semn de întrebare ar trebui să ridice o suspiciune puternică de tetralogie a lui Fallot, în special varianta cu atrezie pulmonară. Acest semn poate fi util pentru screening, considerând că diagnosticul prenatal de TF prin ultrasonografie de rutină rămâne o provocare(10). Constatările ultrasonografice caracteristice TF cu atrezie pulmonară au inclus o mică arteră pulmonară, o aortă mare şi un defect septal ventricular(11).
La fetuşii cu TF apare şi o modificare a axului cardiac. Un ax cardiac anormal este asociat cu prezenţa atreziei pulmonare, a arcului aortic pe partea dreaptă şi cu un risc de deces postnatal mai mare(12).
Ventriculul drept cu dublă cale de ejecţie
Ventriculul drept cu dublă cale de ejecţie (DORV) este o anomalie cardiacă congenitală în care atât aorta, cât şi trunchiul pulmonar apar din ventriculul morfologic drept. DORV reprezintă aproximativ 2% din defectele cardiace congenitale. Este de obicei clasificat ca o anomalie conotruncală şi există aproape întotdeauna un DSV. Pacienţii necesită o varietate de intervenţii chirurgicale, iar prognosticul pe termen lung este variabil. Prin urmare, diagnosticul prenatal este important în a acorda părinţilor o consiliere adecvată(13).
Există mai multe tipuri de DORV. În funcţie de poziţia marilor vase, există DORV cu poziţionarea marilor vase unul lângă celălalt, malpoziţie stângă sau malpoziţie dreaptă a marilor vase. Referitor la localizarea DSV, există DORV cu DSV subaortic, subpulmonar, DORV cu DSV subpulmonar şi subaortic, DORV cu DSV nonsubaortic şi nonsubpulmonar.
Majoritatea feţilor cu DORV pot fi identificaţi antenatal ca având o inimă anormală. Cu toate acestea, este foarte dificil să distingem acest defect particular de alte anomalii conotruncale(14).
În cele mai multe cazuri, o evaluare ecocardiografică fetală atentă poate determina detaliile anatomice esenţiale la feţii cu DORV, pentru a permite o sfătuire exactă cu privire la tipul de intervenţie chirurgicală necesară(13).
Withama a stabilit primele criterii de diagnosticare în 1957, descriind atât aorta, cât şi artera pulmonară care se nasc din ventriculul morfologic drept (VD). Unii autori preferă să utilizeze criteriile mai puţin rigide din Lev şi Anderson, cerând ca un trunchi arterial să se nască complet din VD şi cel puţin jumătate din celălalt trunchi arterial să provină din VD(15).
Utilizarea ecocardiografiei convenţionale este inadecvată pentru diagnosticarea acestei afecţiuni. Câteva studii au arătat beneficiile utilizării cartografierii fluxului color pentru evaluarea defectelor de structură ale inimii fetale, demonstrând avantajul faţă de ecocardiografia convenţională(15).
Diagnosticul prenatal al ventriculului drept cu dublă cale de ejecţie rămâne o provocare ultrasonografică.
În concluzie, deşi există un grad ridicat de precizie în detectarea anomaliilor cardiace fetale, uneori acestea pot fi trecute cu vederea, mai ales în situaţiile unei imagini de patru camere normale la un screening ultrasonografic de rutină.
Dificultăţile în definirea relaţiei spaţiale dintre marile artere reprezintă un factor limitator în diagnosticarea anomaliilor conotruncale în unele cazuri. Poziţia fetală, calitatea imaginii şi vârsta gestaţională târzie pot îngreuna diagnosticul malformaţiilor cardiace conotruncale. În anumite situaţii, aspectul de vase paralele poate duce în mod eronat la diagnosticarea unei malpoziţii de vase mari.
Prognosticul feţilor cu anomalii conotruncale este nefavorabil, chiar şi în absenţa anomaliilor extracardiace majore, cu excepţia feţilor cu tetralogie Fallot.
Acknowledgment: All authors contributed equally to this article.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
1. Vaidyanathan B, Kumar S, Sudhakar A, Krishna Kumar RK. Conotruncal anomalies in the fetus: Referral patterns and pregnancy outcomes in a dedicated fetal cardiology unit in South India. Ann Pediatr Cardiol. 2013;6(1): 15–20.
2. Paladini D, Rustico M, Todros T, Palmieri S, Gaglioti P, Benettoni A, Russo MG, Chiappa E, D'Ottavio G. Conotruncal anomalies in prenatal life. Ultrasound Obstet. Gynecol. 1996;(8): 241-246.
3. Driscoll DA, Salvin J, Sellinger B et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813-817.
4. Paladini D, Pacileo G, Palmieri S et al. Prenatal diagnosis of 22q11 microdeletion in a fetus with a conotruncal heart defect. Ultrasound ObstetGynecol. 1998;11:68-70.
5. Tometzki AJP, Suda K, Kohl T, Kovalchin JP, Silverman NH. Accuracy of prenatal echocardiographic diagnosis and prognosis of fetuses with conotruncal anomalies. Journal of the American College of Cardiology. 1999;33(6).
6. Hamza HS, Gaber KR, Raouf WA, Dohain AM, Mohsen GSA, Abdd Elfattah AN, Senousy SM, Abozid HE, Attia WA. Fetal echocardiography for early detection of conotruncal anomalies in high risk pregnancies: One year follow-up. Journal of Clinical Neonatology 2016. 5(1):35-38
7. Grewal DS, Khanna V, Col, Saxena S, Brig, Chamoli SC. Sonographic diagnosis of transposition of great arteries in mid trimester: Our experience. Med J Armed Forces India. 2016;72(4): 386–388.
8. Gotsch F, Romero R, Espinoza J, Kusanovic JP, Erez O, Hassan S, Yeo L. Prenatal diagnosis of truncus arteriosus using multiplanar display in 4D ultrasonography. J Matern Fetal Neonatal Med. 2010, Apr;23(4): 297–307.
9. Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009; 4: 2.
10. Martínez JM, Gómez O, Bennasar M, Olivella A, Crispi F, Puerto B, Gratacós E. The 'question mark' sign as a new ultrasound marker of tetralogy of Fallot in the fetus. Ultrasound Obstet Gynecol. 2010, Nov;36(5):556-60.
11. Hung JH, Lu JH, Weng ZC, Chen CY, Chao KC, Hung CY. Prenatal diagnosis of tetralogy of Fallot with pulmonary atresia. J Chin Med Assoc. 2008; 71(5):262-6.
12. Zhao Y, Edington S, Fleenor J, Sinkovskaya E, Porche L, Abuhamad A. Fetal cardiac axis in tetralogy of Fallot: associations with prenatal findings, genetic anomalies and postnatal outcome. Ultrasound in Obstetrics and Gynecology. 2016. https://doi.org/10.1002/uog.15998
13. Gelehrter S, Owens ST, Russell MW, van der Velde ME, Gomez-Fifer C. Accuracy of the fetal echocardiogram in double-outlet right ventricle. Congenit Heart Dis. 2007 Jan-Feb;2(1):32-7.
14. Tongsong T, Chanprapaph P, Sittiwangkul R, Khunamornpong S. Antenatal diagnosis of double outlet of right ventricle without extracardiac anomaly: a report of 4 cases. J Clin Ultrasound. 2007, May;35(4):221-5.
15. Augusto L, Chiba Y, Endo S, Ishihara Y. Prenatal Diagnosis of Double Outlet Right Ventricle Using Advanced Dynamic Flow. Journal of Medical Ultrasound. 2003;11 (3): 115-117.
2. Paladini D, Rustico M, Todros T, Palmieri S, Gaglioti P, Benettoni A, Russo MG, Chiappa E, D'Ottavio G. Conotruncal anomalies in prenatal life. Ultrasound Obstet. Gynecol. 1996;(8): 241-246.
3. Driscoll DA, Salvin J, Sellinger B et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813-817.
4. Paladini D, Pacileo G, Palmieri S et al. Prenatal diagnosis of 22q11 microdeletion in a fetus with a conotruncal heart defect. Ultrasound ObstetGynecol. 1998;11:68-70.
5. Tometzki AJP, Suda K, Kohl T, Kovalchin JP, Silverman NH. Accuracy of prenatal echocardiographic diagnosis and prognosis of fetuses with conotruncal anomalies. Journal of the American College of Cardiology. 1999;33(6).
6. Hamza HS, Gaber KR, Raouf WA, Dohain AM, Mohsen GSA, Abdd Elfattah AN, Senousy SM, Abozid HE, Attia WA. Fetal echocardiography for early detection of conotruncal anomalies in high risk pregnancies: One year follow-up. Journal of Clinical Neonatology 2016. 5(1):35-38
7. Grewal DS, Khanna V, Col, Saxena S, Brig, Chamoli SC. Sonographic diagnosis of transposition of great arteries in mid trimester: Our experience. Med J Armed Forces India. 2016;72(4): 386–388.
8. Gotsch F, Romero R, Espinoza J, Kusanovic JP, Erez O, Hassan S, Yeo L. Prenatal diagnosis of truncus arteriosus using multiplanar display in 4D ultrasonography. J Matern Fetal Neonatal Med. 2010, Apr;23(4): 297–307.
9. Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009; 4: 2.
10. Martínez JM, Gómez O, Bennasar M, Olivella A, Crispi F, Puerto B, Gratacós E. The 'question mark' sign as a new ultrasound marker of tetralogy of Fallot in the fetus. Ultrasound Obstet Gynecol. 2010, Nov;36(5):556-60.
11. Hung JH, Lu JH, Weng ZC, Chen CY, Chao KC, Hung CY. Prenatal diagnosis of tetralogy of Fallot with pulmonary atresia. J Chin Med Assoc. 2008; 71(5):262-6.
12. Zhao Y, Edington S, Fleenor J, Sinkovskaya E, Porche L, Abuhamad A. Fetal cardiac axis in tetralogy of Fallot: associations with prenatal findings, genetic anomalies and postnatal outcome. Ultrasound in Obstetrics and Gynecology. 2016. https://doi.org/10.1002/uog.15998
13. Gelehrter S, Owens ST, Russell MW, van der Velde ME, Gomez-Fifer C. Accuracy of the fetal echocardiogram in double-outlet right ventricle. Congenit Heart Dis. 2007 Jan-Feb;2(1):32-7.
14. Tongsong T, Chanprapaph P, Sittiwangkul R, Khunamornpong S. Antenatal diagnosis of double outlet of right ventricle without extracardiac anomaly: a report of 4 cases. J Clin Ultrasound. 2007, May;35(4):221-5.
15. Augusto L, Chiba Y, Endo S, Ishihara Y. Prenatal Diagnosis of Double Outlet Right Ventricle Using Advanced Dynamic Flow. Journal of Medical Ultrasound. 2003;11 (3): 115-117.