Incontinentia pigmenti, known as Bloch-Sulzberger disease, is a rare genetic disorder with an incidence of about 1 in 40000 newborns. The first cases were reported by dermatologists Bruno Bloch in 1926 and Marion Sulzberger in 1928. Incontinentia pigmenti occurs by inactivating a dominant X-linked IKBKG (NEMO) gene responsible for the regulation of the Kappa B nuclear factor. The Kappa B nuclear factor complex is responsible for protecting cells against self-destruction. Affected tissues are those with ectodermal origin: teeth, hair, eyes, central nervous system. For the male fetus, spontaneous abortion occurs most often. Feminine newborn babies may be healthy or manifest. We present a case of incontinentia pigmenti in a newborn female with skin and neurological manifestations from birth.
Incontinentia pigmenti
Incontinentia pigmenti
First published: 23 iunie 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.2.2.2018.1814
Abstract
Rezumat
Incontinentia pigmenti, cunoscută ca boala Bloch-Sulzberger, reprezintă o afecţiune genetică rară în lume, având o incidenţă de aproximativ 1 la 40000 de nou-născuţi. Primele cazuri au fost raportate de medicii dermatologi Bruno Bloch în 1926, respectiv Marion Sulzberger în 1928. Incontinentia pigmenti apare prin inactivarea unei gene dominante X-linkate, IKBKG (NEMO), responsabilă de reglarea factorului nuclear Kappa B. Complexul de factori nuclear Kappa B este responsabil de protejarea celulelor de a nu se autodistruge. Ţesuturile afectate sunt cele cu origine ectodermală: dinţi, păr, ochi, sistem nervos central. La fătul de sex masculin, intervine de cele mai multe ori avortul spontan. Nou-născuţii de sex feminin pot să fie purtători sănătoşi sau manifeşti. Prezentăm un caz de incontinentia pigmenti la un nou-născut de sex feminin, cu manifestări cutanate şi neurologice de la naştere.
Introducere
Incontinentia pigmenti, sau boala Bloch-Sulzberger, este o afecţiune multisistemică X-linkată dominantă. Copiii de sex feminin sunt purtătorii acestei inactivări ale genei IKBKG/NEMO şi prezintă afectare multiorganică mai mare sau mai mică, în funcţie de severitatea afectării genice.
Din punct de vedere clinic şi morfopatologic incontinentia pigmenti prezintă patru stadii (tabelul 1).
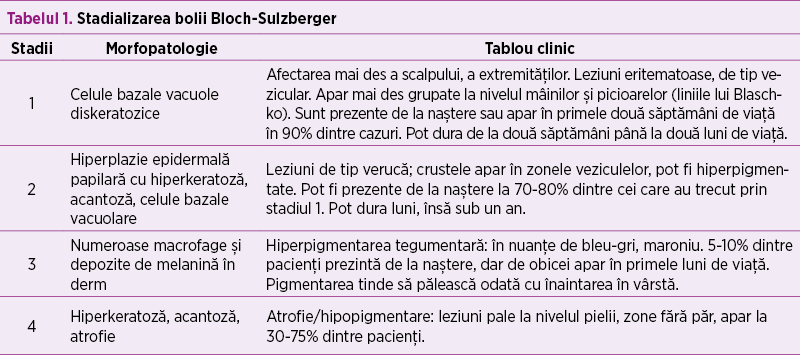
Alte organe afectate sunt dinţii (erupţii întârziate, modificări de formă), fanere (modificări de formă ale unghiilor), păr (pierderi sau lipsă de păr, absenţa sprâncenelor şi a genelor), oculare (apar la 20-35% dintre pacienţi, înainte de vârsta de 5 ani, pot da cecitate dacă nu se intervine precoce), sistem nervos central (convulsii în primele săptămâni de viaţă, retard de dezvoltare neuromotorie, paralizie spastică, atrofie cerebrală), anomalii scheletale, anomalii pulmonare şi cardiace.
Investigaţiile de laborator cuprind biopsia de piele, testarea prenatală, CT/IRM, teste de sânge, angiografie cu fluorosceină, examen oftalmologic.
Diagnostic diferenţial: sindrom MIDAS, hipomelanoză ITO, hipoplazie dermală GOLTZ, sindrom NAEGELI.
Prezentare de caz
Un copil de sex feminin, A.T., provenit din sarcină dispensarizată, GVI PII (patru avorturi spontane), cu restricţie de creştere intrauterină de două săptămâni, extrasă prin operaţie cezariană, din prezentaţie craniană, SA 9, a prezentat leziuni pustuloase la nivel cefalic, al trunchiului şi membrelor, descuamări, retrognatism, torticolis latero-cervical stâng poziţional, echilibrat cardiopulmonar, tonus normal. Existenţa leziunilor tegumentare a ridicat suspiciunea unei infecţii materno-fetale, motiv pentru care s-a iniţiat tratament cu Cefort® până la infirmarea riscului infecţios (hemograme şi proteina C reactivă normale în dinamică, tratament menţinut 72 de ore). Clinic, nou-născutul a supt cu dificultate la sân şi a primit completare. Din ziua a patra, nou-născutul a început să prezinte discrete mioclonii (valorile glicemiei normale) intermitente la manipulare, cu tonus axial diminuat, simptomatologie care s-a accentuat pe parcursul următoarelor două zile, cu degradarea stării generale, echilibrat din punct de vedere hemodinamic şi cardiopulmonar, dar a primit din ce în ce mai greu alimentaţia la seringă/biberon şi sân, având o curbă ponderală descendentă. S-a transferat în compartimentul de terapie intensivă neonatală şi a primit perfuzie endovenoasă cu soluţie de glucoză 10%, aproximativ 24 de ore, alimentaţie cu formulă la biberon, fiind monitorizat cardiorespirator, cu reluarea creşterii ponderale lent. La 9 zile de viaţă a prezentat abdomen uşor destins de volum, cu contur discret de anse, scaun cu mucozităţi. S-a efectuat coprocultura, care a evidenţiat prezenţa bacteriei Klebsiella. De menţionat că simptomatologia s-a remis spontan până la aflarea rezultatelor. Ţinând cont de starea copilului, s-a decis tratarea conform antibiogramei (ciprofloxacină 20 mg/kgc/zi, 7 zile) cu negativarea probei de control.
Pe parcursul spitalizării a prezentat puseuri eruptive sub forma unor placarde eritematoase care au prezentat ameliorare sub administrare de dexametazonă. Consultul dermatologic a fost efectuat pe 4.01.2016 în Spitalul CF2, fiind diagnosticată cu eczemă atopică şi primind recomandarea de a utiliza Atoderm®.
De asemenea, intermitent, a prezentat un suflu sistolic de grad I-II/6 parasternal stâng.
S-a solicitat şi s-a efectuat consult neurologic, care a constatat uşoară asimetrie a reflexelor arhaice şi diminuarea mişcărilor active ale membrelor inferioare. La ecografia transfontanelară s-au evidenţiat zone hiperecogene cu diametru de 15 mm la nivelul ganglionilor bazali dreapta şi 7 mm la nivelul ganglionilor bazali stânga. S-a efectuat examen IRM, care a evidenţiat leziuni cerebrale multiple cu un numitor comun „stroke-like”, cu posibilă întrerupere de fibre piramidale pe partea dreaptă.
S-a externat prin transfer către Clinica de neurologie a Spitalului Clinic de Copii „V. Gomoiu” Bucureşti.
În evoluţie, copilul a fost monitorizat în Clinica de neurologie a Spitalului Clinic de Copii „V. Gomoiu”, unde s-a stabilit şi diagnosticul de incontinentia pigmenti. Leziunile tegumentare au prezentat ameliorare după vârsta de 3 luni. La 5 luni, copilul a prezentat spasme infantile, necesitând repetate internări, şi a primit tratament cu ACTH, cu evoluţie lent favorabilă. De asemenea, s-a suspicionat diagnosticul de cardiomiopatie hipertrofică, însă a fost infirmat în timp. Pentru luxaţia congenitală de şold s-au încercat diverse sisteme de tip ham, intervenţia chirurgicală nefiind o soluţie în prezenţa leziunilor tegumentare. Consultul oftalmologic a stabilit vicii de refracţie, fiind exclusă afectarea retiniană. Din punct de vedere neurologic, copilul urmează terapii diverse de tip Bowen, NACD, metoda Anat Baniel, în urma cărora recuperarea este favorabilă, dar lentă; imaginile cerebrale IRM au evidenţiat ţesut cicatricial, fără alte zone noi de infarctizare şi cu remisiunea episoadelor de convulsii. În cei doi ani, mama copilului a fost la rândul ei diagnosticată cu incontinentia pigmenti pe baza datelor medicale de la naşterea acesteia (a fost internată în primele luni de la naştere la INSMC „Alessandrescu-Rusescu” pentru o formă severă de dermatită, alopecie, probleme de dentiţie şi de vedere, ulterior).
Concluzie
Un nou-născut este întotdeauna o provocare. Eritemul unui nou-născut poate să reprezinte mai mult decât un incident fiziologic. Când elementele cutanate au o evoluţie atipică, este important să acordăm atenţie tuturor semnelor indirecte ale copilului, precum şi datelor anamnestice materne.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
2. Sahin M, Neurocutaneous syndromes. In: Kliegman R, Stanton B, St Geme J, Schor N, Nelson. Textbook of Pediatrics, 20 Edition. Elsevier. 2015: 2881-2882.
3. Agarwal P. Incontinentia pigmenti. Indian J Dermatol Venereol Leprol. 1997; 63(6):368-9.
4. El Fekih L, Hmaied W, Souissi K, Nasri H, Derbel F, Hamdi A. Incontinentia pigmenti: a rare cause of retinal vasculitis in children. Tunis Med. 2008; 86(12):1079-81.
5. Conte MI, Pescatore A, Paciolla M, Esposito E, Milano MG et al. Insight into IKBKG/ NEMO locus: report of new mutations and complex genomic rearrangements leading to incontinentia pigmenti disease. Human Mutation [Hum Mutat]. 2014; Feb; Vol.35 (2),pp 165-77.
6. Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol. 2004; Jun;140(6):748-50. Review.
7. Minic S, Trpinac D, Obradovic M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet Journal of Rare Disease. 2013;(8):25.
8. Bruckner AL. Incontinentia pigmenti: a window to the role of NF-kappaB function. Semin Cutan Med Surg. 2004; Jun;23(2):116-24. Review.
9. https://ghr.nlm.nih.gov/condition/incontinentia-pigmenti#sourcesforpage