Omphalocele is the most common congenital parietal defect, with an incidence of 1 to 4,000-5,000 live births. Depending on the contents of the hernial sac, the omphalocele may be of low parietal defect when the extracorporeal sac contains only stomach and bowels, or with high parietal defects, when the extracorporeal sac contains the liver. Published data over time have shown an important association between parietal defect and chromosomal anomalies, so that after ultrasound detection of omphalocele there are required a fetal cardiac ultrasound and amniocentesis. The most common chromosomal abnormalities are: Trisomy 18, Trisomy 13, triploidy, Trisomy 21, 45X, 47XXY and 47XXX, partial aneuploidy - deletions, duplications, inversions. The risk of chromosomal anomalies varies with maternal age, gestational age, umbilical cord cyst association, omphalocele sac content, complexity of other associated abnormalities. In fetuses with ventral parietal defects, an important role will be played by the detection of other associated defects and karyotyping in order to determine the course of pregnancy. Early involvement of the neonatologist and pediatric surgeon can improve perinatal and postnatal prognosis in fetuses with simple parietal defects without chromosomal abnormalities.
Omfalocelul - asocierea cu anomalii cromozomiale şi abord terapeutic
Omphalocele - association with chromosomal abnormalities and therapeutic approach
First published: 16 iunie 2017
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.1.2.2017.830
Abstract
Rezumat
O mfalocelul reprezintă cel mai des întâlnit defect parietal congenital, având o incidenţă de 1 la 4000-5000 de naşterii vii. În funcţie de conţinutul sacului, omfalocelul poate fi cu defect parietal mic, atunci când sacul extracorporeal conţine doar stomacul şi intestinele, sau cu defect parietal mare, atunci când sacul extracorporeal conţine şi ficatul. Datele publicate de-a lungul timpului au arătat o importantă asociere între defectul parietal şi anomaliile cromozomiale, astfel încât după depistarea ecografică a omfalocelului este necesară efectuarea unei ecografii cardiace fetale şi a amniocentezei. Anomaliile cromozomiale cel mai des întâlnite sunt: trisomia 18, trisomia 13, triploidia, trisomia 21, 45X, 47XXY şi 47XXX, aneuploidiile parţiale – deleţii, duplicaţii, inversiuni. Riscul anomaliilor cromozomiale variază cu vârsta maternă, vârsta gestaţională, asocierea cu chisturi ale cordonului ombilical, conţinutul sacului omfalocelului, complexitatea altor anomalii asociate. În cazul feţilor cu defecte parietale ventrale, un rol important îl vor juca detectarea altor defecte asociate şi cariotiparea, în vederea stabilirii cursului sarcinii. Implicarea cât mai precoce a neonatologului şi a chirurgului pediatric poate îmbunătăţi prognosticul perinatal şi postnatal la feţii cu simple defecte parietale, fără anomalii cromozomiale.
Introducere
Omfalocelul a fost descris prima dată de Ambrose Paré în 1634 şi, deşi reprezintă o patologie relativ rară, având o incidenţă de 1 la 4000-5000 de feţi născuţi vii, este totuşi cea mai frecventă anomalie de perete ventral.Se defineşte ca un defect de linie mediană a peretelui abdominal anterior situat la baza de inserare a cordonului ombilical, manifestat prin hernierea unor viscere abdominale într-un sac herniar avascular. Prognosticul acestui defect depinde în cea mai mare măsură de asocierea lui cu alte anomalii structurale sau cromozomiale(1).
Patogeneză
Dezvoltarea tractului digestiv începe încă din săptămânile a 3-a şi a 4-a, odată cu plicaturarea longitudinală şi laterală a embrionului. Intestinul primitiv se formează din endoderm şi este împărţit în proenteron - din care se vor dezvolta faringele, tractul respirator inferior, esofagul şi stomacul, duodenul, ficatul, ductele biliare şi pancreasul; mezenteron - din care se vor dezvolta intestinul subţire, cecul, apendicele cecal, colonul ascendent şi o parte din colonul transvers; şi metenteron - din care se vor forma colonul transvers, descendent, sigmoidul, rectul, canalul anal, o porţiune din vezica urinară şi uretră(2).Mezenteronul, sau intestinul mijlociu, păstrează temporar comunicarea cu sacul vitelin prin ductul vitelin (ductul omfalomezenteric). Începând cu a şasea săptămână de gestaţie, apare o elongaţie a mezenteronului sub forma literei U, care depăşeşte corpul fetal prin ductul omfalomezenteric - acest fenomen fiind descris ca herniere fiziologică prin cordonul ombilical (figura 1)(2,3).
La 10 săptămâni de gestaţie, acest fenomen regresează în mod fiziologic, iar intestinele revin în corpul embrionar, peretele abdominal ventral se închide, iar ductul omfalomezenteric se contractă, formând inserţia cordonului ombilical(3).
Cu toate că etiopatogenia apariţiei omfalocelului nu este pe deplin cunoscută, acesta este determinat de o tulburare a mecanismului de închidere a corpului embrionar, cu hernierea unor visecere în afara cavităţii abdominale prin inelul ombilical, acoperite de o membrană, avasculară, transparentă(4).
La momentul actual sunt acceptate trei teorii ale formării omfalocelului: I - dezvoltare incompletă sau migrare incompletă a faldurilor abdominale; II - persistenţa pediculului omfalomezenteric; III - eşecul viscerelor abdominale de a se întoarce în cavitatea abdominală la sfârşitul celei de-a 10 săptămâni a vieţii intrauterine, după hernierea fiziologică ombilicală(5,6,7).
Majoritatea omfalocelelor sunt centrale şi se formează printr-un defect de plicaturare laterală; în cazuri mai rare, omfalocelul poate fi situat deasupra ombilicului sau dedesubtul acestuia.
Omfalocelul epigastric se formează printr-un defect al faldului cranial şi se asociază unui sindrom de linie mediană superioară - pentalogia Cantrell (sindrom Cantrell-Heller-Ravitch) -, defect parietal, hernie diafragmatică, cord ectopic, lipsa pericardului, cleft sternal şi defecte cardiace congenitale - sindrom extrem de rar, circa 5 cazuri la 1 milion de naşteri; până la momentul actual sunt raportate aproximativ 90 de cazuri în literatura de specialitate(8,9).
Omfalocelul hipogastric se asociază cu anomalii ale enteronului terminal: extrofie vezicală, atrezie colonică, mielomeningocel, anomalii ale vertebrelor sacrate(8,10).
Sacul herniar al omfalocelului poate conţine: intestin subţire, intestin gros, stomac, ficat, splină, vezică urinară şi chiar gonade(11). În circa 50% dintre cazuri, ficatul este extracorporeal(10,11).
Diametrul defectului de perete variază, în general, între 4 şi 12 cm - un diametru >8 cm este însoţit de prezenţa ficatului extracorporeal(11). În aproximativ 10-18% dintre cazuri, poate să apară o ruptură a sacului herniar al omfalocelului înainte de naştere(4). Un omfalocel este considerat gigant atunci când defectul parietal este mai mare de 5 cm. În aceste cazuri, cavitatea abdominală rămâne mică şi subdezvoltată(4,5,11).
Epidemiologie
Datele epidemiologice arată o asociere cu vârsta maternă crescută, o frecvenţă mai mare la feţii de sex masculin (1,5-3: 1), apariţia la gemeni, la naşteri multiple, la diferite generaţii din cadrul aceleiaşi familii şi o asociere cu trisomiile 13, 18 şi 21 (25-50% dintre cazuri) şi cu sindromul Beckwith-Wiedemann, motiv pentru care fost emisă şi o teorie genetică pentru omfalocel(4,12,13,14).Asocieri
Aşadar, în 30-80% dintre cazuri, omfalocelul poate asocia alte anomalii, după cum urmează: anomali cardiace în 50% dintre cazuri (tetralogie Fallot şi/sau defecte septale atriale); anomalii cromozomiale în 30-40% dintre cazuri (cel mai frecvent, trisomie 13 şi 18 şi sindrom Beckwith-Wiedemann - macrosomie, hipoglicemie şi macroglosie); anomalii gastrointestinale în 40% dintre cazuri şi în mai puţin de 10% dintre cazuri poate asocia anomalii genitourinare, faciale, scheletale sau renale(8,11,14,15,16). Au fost raportate, de asemenea, cazuri de trisomie: 14, 15, 16, 17, precum şi cazuri de triploidii(15). Anomaliile cromozomiale însoţesc mai frecvent defectele parietale mici, în timp ce defectele parietale mari sunt complicate cu alte anomalii structurale, cel mai frecvent cardiace sau hipoplazie pulmonară(17,18,19).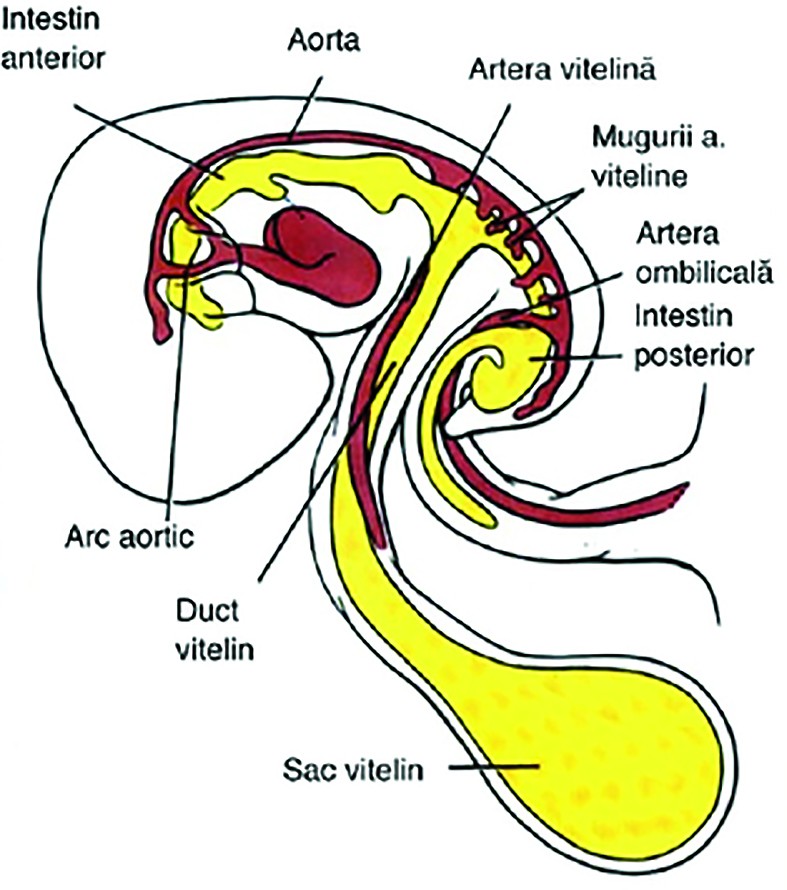
Management
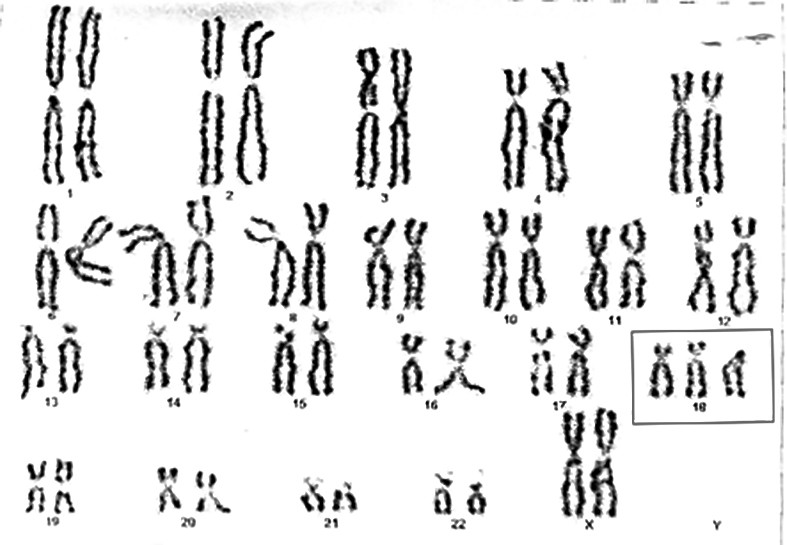
Un făt cu omfalocel trebuie evaluat atât din punct de vedere structural - din cauza asocierilor cu alte defecte structurale -, cât şi din punct de vedere genetic, cunoscut fiind faptul că poate coexista şi o aberaţie cromozomială. Din acest motiv, ecografia de morfologie fetală ocupă un loc important, la fel ca ecocardiografia fetală - malformaţiile cardiace fiind cel mai frecvent asociate. Amniocenteza cu efectuarea cariotipării fetale este, de asemenea, o evaluare obligatorie. După stabilirea unui diagnostic complet, se va efectua consilierea genetică, acolo unde este cazul. După efectuarea unei informări complete asupra diagnosticului, prognosticului şi posibilelor complicaţii, părinţii pot stabili dacă doresc sau nu continuarea cursului sarcinii până în 24 de săptămâni, în termen legal.
Datele din literatură arată că circa 29-51% dintre părinţi solicită întreruperea cursului sarcinii(13,20,21). Atunci când se decide totuşi continuarea cursului sarcinii, sunt necesare ecografii seriate pentru monitorizarea stării de sănătate a fătului - până la 35% dintre cazuri pot dezvolta restricţie de creştere intrauterină(12,15).
Naşterea trebuie programată într-un serviciu specializat, de obicei o maternitate de gradul 3, iar ulterior nou-născutul trebuie preluat de un serviciu de chirurgie pediatrică în vederea reparării defectului parietal. Este de preferat naşterea prin operaţie cezariană, pentru a proteja sacul herniar şi pentru a evita posibilele distocii din timpul unui travaliu, accidente hemoragice - ruptură hepatică fetală, atunci când ficatul este componentă a sacului herniar, sau accidente infecţioase - cunoscut fiind faptul că tractul genital matern este populat cu diverşi germeni. Totuşi, naşterea pe cale vaginală poate fi încercată la feţii cu defect parietal mic.
Imediat postnatal sunt importante susţinerea cardiorespiratorie a nou-născutului, protejarea sacului herniar - comprese umede, asigurarea homeostaziei, a normotermiei - şi transferul cât mai rapid într-un serviciu de chirurgie pediatrică.
Ratele de supravieţuire sunt direct influenţate de anomalii cromozomiale sau structurale asociate. Mortalitatea poate atinge 80% la feţii care asociază alte anomalii(15). De asemenea, un factor de prognostic este dat şi de dimensiunea sacului herniar, mortalitatea fiind mai crescută la omfalocelele mari, prin detresă repiratorie, complicaţii infecţioase sau insuficienţă hepatică. Rata de supravieţuire poate atinge 95% la cazurile de omfalocel mic şi fără alte asocieri(4,15,18,22,23).
În loc de concluzii, vă propunem o prezentare de caz în imagini
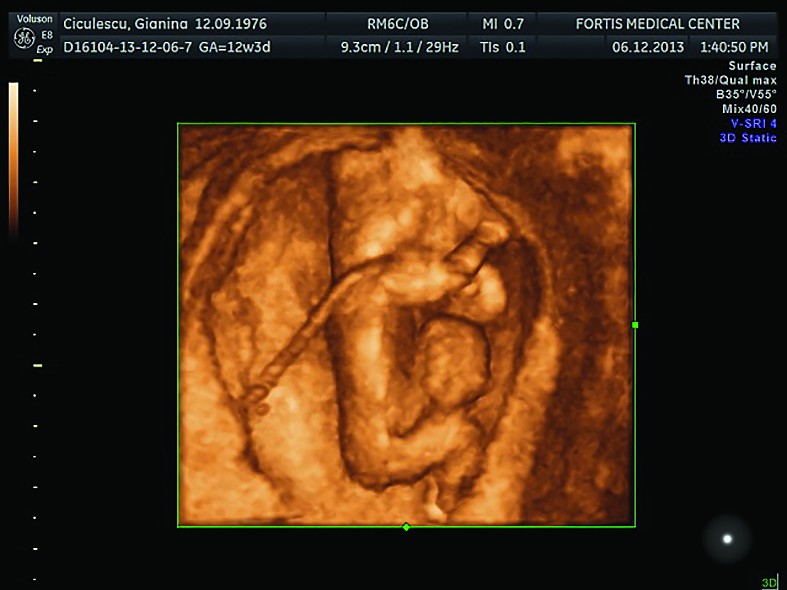
Cazul 1:
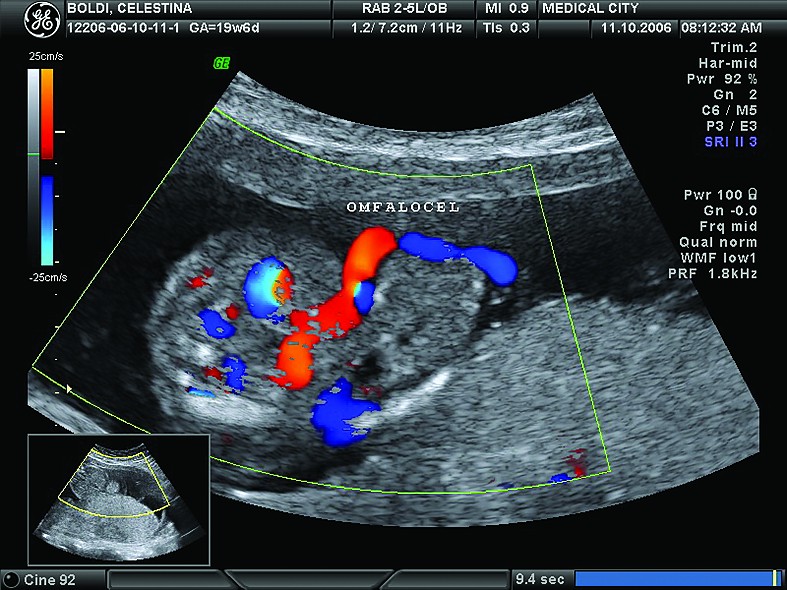
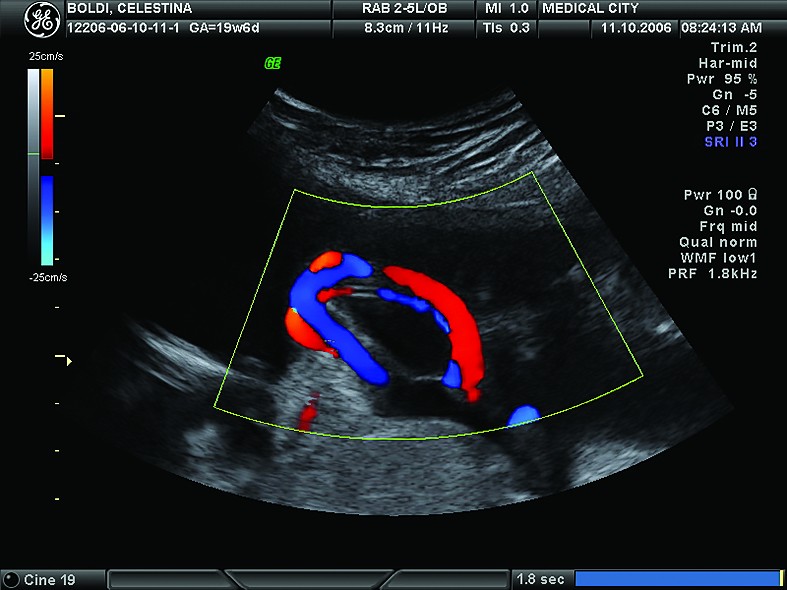
Cazul 2:
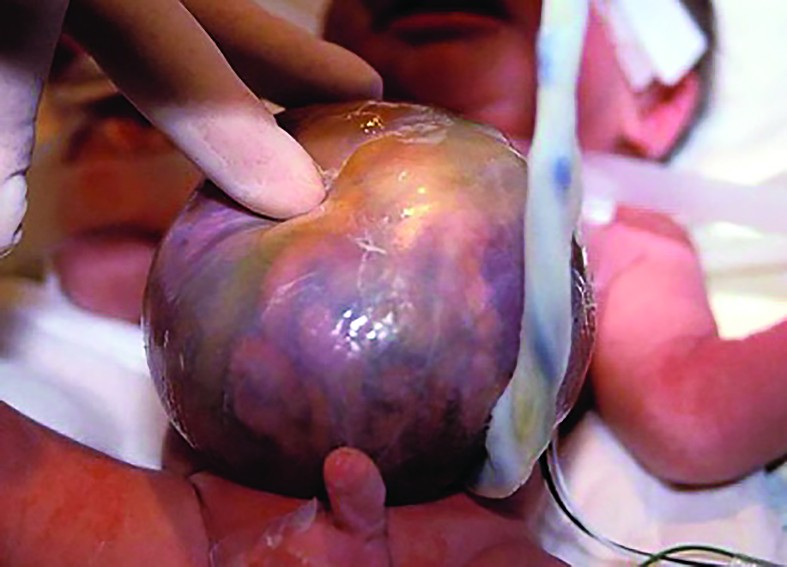
Bibliografie
2. Moore K, and Persaud T. 1998. In The Developing Human Clinically Oriented Embryology, 6th ed., Moore K, and Persaud T, eds. Philadelphia: WB Saunders, 273–302.
3. Seashore JH. 1978. Congenital abdominal wall defects. Clinics in Perinatology 5(1): 61–77.
4. Cooney DR. 1998. Defects of the abdominal wall. In Pediatric Surgery, 5th ed., O’Neill JA, et al., eds. Toronto: Mosby, 1045–1086.
5. Dillon PW, and Cilley RE. 1993. Newborn surgical emergencies: Gastrointestinal anomalies, abdominal wall defects. Pediatric Clinics of North America 40(6): 1289–1314.
6. Sadler T. 2000. Digestive system. In Langman’s Medical Embryology, 8th ed., O’Brian P, and Sadler T, eds. Philadelphia: Lippincott Williams & Wilkins, 270–303.
7. Rescorla FJ. 2001. Surgical emergenices in the newborn. In Workbook in Practical Neonatology, 3rd ed., Polin RA, Yoder MC, and Burg FD, eds. Toronto: WB Saunders, 423–459.
8. Wilson RD, and Johnson MP. 2004. Congenital abdominal wall defects: An update. Fetal Diagnosis and Therapy 19(5): 385–398.
9. Lack, Vered, et al. Pentalogy of Cantrell with thoracoabdominal ectopia cordis: Attempted surgical correction and review of recent literature to aid prognostication prior to surgery. Journal of Pediatric Surgery Case Reports 3.11 (2015): 476-480.
10. Grosfeld JL, Dawes L, and Weber TR. 1981. Congenital abdominal wall defects: Current management and survival. Surgical Clinics of North America 61(5): 1037–1049.
11. Dykes EH. 1996. Prenatal diagnosis and management of abdominal wall defects. Seminars in Pediatric Surgery 5(2): 90–94.
12. Paidas MJ, Crombleholme TM, and Robertson FM. 1994. Prenatal diagnosis and management of the fetus with an abdominal wall defect. Seminars in Perinatology 18(3): 196–214.
13. Forrester MB, and Merz RD. 1999. Epidemiology of abdominal wall defects, Hawaii, 1986–1997. Teratology 60(3): 117–123.
14. Calzolari E, et al. 1995. Omphalocele and gastroschisis in Europe: A survey of 3 million births 1980–1990. EUROCAT Working Group. American Journal of Medical Genetics 58(2): 187–194.
15. Heider AL, Strauss RA, and Kuller JA. 2004. Omphalocele: Clinical outcomes in cases with normal karyotypes. American Journal of Obstetrics and Gynecology 190(1): 135–141.
16. Nyberg DA, et al. 1989. Chromosomal abnormalities in fetuses with omphalocele. Significance of omphalocele contents. Journal of Ultrasound Medicine 8(6): 299–308.
17. Yang P, et al. 1992. Genetic-epidemiologic study of omphalocele and gastroschisis: evidence for heterogeneity. American Journal of Medical Genetics 44(5): 668–675.
18. Biard J, et al. 2004. Prenatally diagnosed giant omphaloceles: Short- and long-term outcomes. Prenatal Diagnosis 24(6): 434–439.
19. Hershenson MB, et al. 1985. Respiratory insufficiency in newborns with abdominal wall defects. Journal of Pediatric Surgery 20(4): 348–353.
20. Stoll C, et al. 2001. Risk factors in congenital abdominal wall defects (omphalocele and gastroschisis): A study in a series of 265,858 consecutive births. Annals of Genetics 44(4): 201–208.
21. Barisic I, et al. 2001. Evaluation of prenatal ultrasound diagnosis of fetal abdominal wall defects by 19 European registries. Ultrasound in Obstetrics & Gynecology 18(4): 309–316.
22. Yaster M, et al. 1988. Hemodynamic effects of primary closure of omphalocele/gastroschisis in human newborns. Anesthesiology 69(1): 84–88.
23. Fisher R, et al. 1996. Impact of antenatal diagnosis on incidence and prognosis in abdominal wall defects. Journal of Pediatric Surgery 31(4): 538–541.