Distrofia musculară progresivă - update
Duchenne muscular dystrophy - update
Abstract
Duchenne muscular dystrophy (DMD) is a progressive, severely disabling neuromuscular disease. In Duchenne muscular dystrophy, the open reading frame of the X-linked dystrophin gene (DMD) is disrupted by deletions (roughly 65%), duplications (10%), point mutations (10%) or other smaller rearrangements. If untreated, loss of ambulation is typically seen in 10-12 years. Death occurs in early to mid-twenties due to heart failure. It has been searching for alternative therapeutic approaches for patients with muscular dystrophies. New advances in the management of DMD use exon skipping, gene therapy and cellular therapy seems to slow its progression. We report the case of a 6-year old boy with DMD and new research in DMD treatment.Keywords
Duchenne muscular dystrophychildrenRezumat
Distrofia musculară Duchenne (DMD) este o afecțiune neuromusculară progresivă, debilitantă. În DMD, gena distrofinei este afectată de deleție (aproximativ 65%), duplicație (10%) sau alte mutații. Fără tratament, pierderea mersului se produce între 10 și 12 ani. Decesul survine rapid, în jurul vârstei de 20 de ani, prin insuficiență cardiacă. S-au încercat o serie de abordări terapeutice pentru acești pacienți. Noile descoperiri privind utilizarea „exon skipping”, a terapiei genice și celulare, se pare că îi încetinesc progresia. Prezentăm cazul unui pacient în vârstă de 6 ani cu DMD, precum și noi cercetări în tratamentul acestei afecțiuni.Cuvinte Cheie
distrofie musculară DuchennecopiiIntroducere
Distrofia musculară progresivă Duchenne a fost descrisă de Guillaume Duchenne de Boulogne în 1868. Este forma clinică cea mai comună la copil, incidența bolii fiind de 1 caz/3.000-3.500 de nou-născuți de sex masculin. Este o boală condiționată genetic, transmisă X-linkat, prin urmare toate cazurile sunt totdeauna băieți. Excepțional poate apărea fenotip feminin: sindrom Turner (45, XO) în care cromozomul X este purtător al tarei, fete purtătoare a unei inversiuni a cromozomului X sau a unei translocații X autozomale (cromozomul X normal este inactivat, iar cromozomul X care a suferit translocația este singurul funcțional). 75% din cazurile de DMD se produc prin mutații noi! Femeia este tipic purtătoare a tarei, dar află acest lucru doar în momentul în care are un fiu afectat; fiul mamei purtătoare are șansa de 50% să moștenească gena defectivă de la mamă; fiica mamei purtătoare are 50% șansă de a fi purtătoare și 50% șansă de a avea două copii normale ale genei(1).
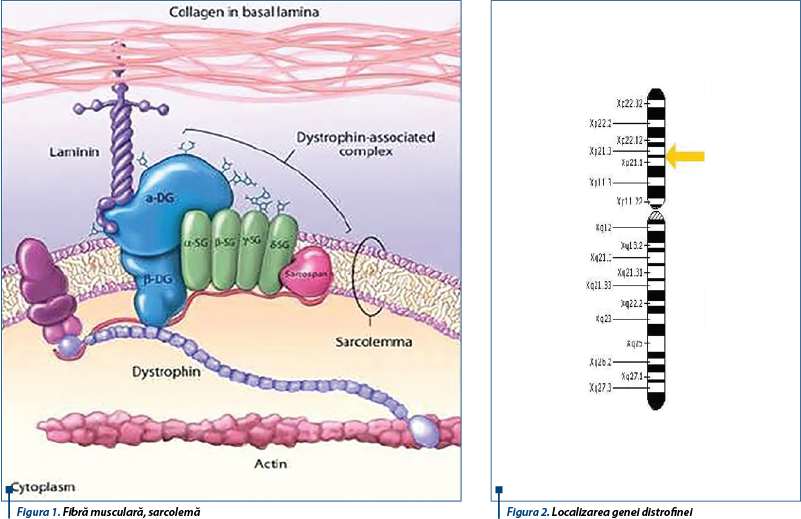
Proteina deficitară din DMD este DISTROFINA, proteina responsabilă de conectarea citoscheletului fibrelor musculare la matrixul extracelular, ceea ce permite menținerea stabilității fibrelor musculare în contracție (figura 1). Absența distrofinei permite pătrunderea calciului în exces prin sarcolemă, ceea ce duce la acumularea apei în exces în mitocondrii, care sunt distruse. În mușchiul distrofic, această disfuncționalitate a mitocondriei duce la creșterea amplificării stresului oxidativ, în final la moarte celulară și înlocuirea fibrelor musculare cu țesut adipos și țesut conjunctiv. Distrofina se mai găsește, în afară de mușchii scheletici, în miocard, creier și retină. Gena distrofinei este localizată pe brațul scurt al cromozomului X (banda p21) (figura 2); este una din cele mai mari gene umane; la acest nivel se pot produce diverse mutații: deleții, duplicații, mutații punctiforme(2).
Clinic, boala este prezentă de la naștere, dar bolnavul devine manifest clinic între 3 și 5 ani, prin afectarea proximală a mușchilor, cu scăderea progresivă a forței și masei musculare, cu dificultăți în alergare, în urcatul scărilor, căderi anormal de frecvente, cu dificultăți de ridicare, aspect evidențiat clinic prin manevra Gowers pozitivă. Scăderea forței musculare la membrele inferioare și la nivelul bazinului determină tulburări de mers, „mers de rață” și deformări osoase - lordoză lombară. Afectarea musculaturii membrelor superioare este discretă, bilaterală, ușor asimetrică, deficitul motor este aparent; flexorii gâtului sunt afectați precoce (SCMI), iar imposibilitatea flexiei gâtului în clinostatism diferențiază DMD de distrofia musculară Becker. Fața rămâne indemnă până în stadiile finale ale bolii. Hipertrofia musculară imprimă aspect atletic al moleților, cvadricepșilor, ulterior al centurii scapulare (deltoid și triceps brahial). Creșterea de volum a musculaturii poartă denumirea de pseudohipertrofie deoarece mușchiul normal este înlocuit cu depozite de țesut conjunctiv și grăsimi; la palpare, acești mușchi au fermitate excesivă. În jurul vârstei de 6-7 ani apar contractura tendonului lui Achile, atrofii ale musculaturii pelviene și sacrolombare. Scolioza progresivă și deformarea toracică duc la alterarea dinamicii respiratorii deja reduse de slăbiciunea musculară. Reflexele rotuliene pot fi abolite precoce, iar cele achiliene se pot menține normale mult timp. Afectarea cardiacă apare la 95% din bolnavi, precoce fiind tulburările de ritm și de conducere, iar tardiv, cardiomiopatia (deoarece distrofina se găsește și în mușchiul cardiac). De asemenea, tulburările neuropsihiatrice sunt constante, IQ-ul este scăzut, apar tulburări de comportament. La 12 ani, cei mai mulți pacienți sunt imobilizați în scaun cu rotile. Biologic, enzimele musculare sunt crescute - TGO, TGP, CPK, LDH, aldolaza (20-100xN). Creșterea CPK >1000 UI/L este sugestivă pentru boală. EMG are aspect de traseu miopatic. Biopsia musculară este sugestivă. Diagnosticul este confirmat prin evidențierea deficitului de distrofină (pe biopsia musculară) prin metode imunohistochimice (utilizarea de Ac monoclonali) și genetic prin analiza mutațiilor genetice. Diagnosticul prenatal se efectuează prin analiza AND-ului genomic din celulele amniotice sau vilozitățile coriale prelevate prin amniocenteză după săptămâna a 15-a de sarcină.
În ultimii 5 ani (ianuarie 2012 - noiembrie 2016) s-au prezentat în Clinica II Pediatrie a Spitalului Clinic de Urgenţă pentru Copii „Sfânta Maria” Iaşi 28 de pacienți, cu vârsta cuprinsă între 7 luni și 16 ani, diagnosticați cu miopatie. Dintre cei 28 de pacienți, 26 sunt de sex masculin, iar 2 pacienți de sex feminin. Din acest bilanț s-au exclus pacienții la care afectarea musculară a fost de cauză infecțioasă, metabolică, autoimună sau medicamentoasă.
Dintre cei 28 de pacienți, 13 au fost diagnosticați cu distrofie musculară Duchenne - DMD (diagnostic confirmat prin biopsie musculară sugestivă/examen genetic), 7 cu distrofie musculară Becker, iar 8 nu au fost încadrați într-o miopatie.
Pacienții diagnosticați cu DMD, toți băieți, au vârste cuprinse între 7 luni și 14 ani, cei cu DM Becker - între 6 și 16 ani, iar cei cu miopatie neclasificată au vârste cuprinse între 1 și 11 ani; cele două paciente provin ambele din acest ultim grup.
Caz clinic
Detaliem mai jos cazul unui pacient de 6 ani și jumătate, P.R., din mediu rural, diagnosticat de 4 luni cu DMD în Clinica II Pediatrie. Antecedentele heredo-familiale relevă faptul că este singurul copil al unui cuplu tânăr (mama - 30 de ani, tatăl - 34 de ani), aparent sănătoși, fără boli cronice, autoimune sau musculare în familie. Antecedentele personale fiziologice evidențiază că a fost născut natural, la termen, cu o greutate la naștere de 3.200 g, Apgar 9, nu a prezentat icter fiziologic, alimentat exclusiv natural 9 luni, diversificat tardiv și incorect, vaccinat conform schemei naționale (declarativ), dar profilactizat incorect pentru rahitismul carențial (doar până la un an, cu câte 5 picături de vigantol/zi). Antecedentele personale patologice și istoricul bolii relevă faptul că la vârsta de 3 luni prezintă un traumatism craniocerebral (TCC) prin cădere din pat, la domiciliu, în zilele următoare sugarul prezentând agitație și vărsături, pentru care mama se adresează (după 3 zile de la eveniment) Spitalului „Sf. Maria” - Secția de Neurochirurgie. Este evaluat pentru TTC prin computer tomografie (CT) craniocerebrală, se decelează un important hematom parietal, subiacent - decolare de volet parietal, iar biologic, o anemie ușoară (Hb = 10,43 mg/dl) normocromă, normocitară și un sindrom de citoliză hepatică ~12xN (TGP = 298 U/L, TGO = 356U/L).
O lună mai târziu, o nouă internare de control în Secția de Neurochirurgie relevă aceeași citoliză hepatică (TGP = 258 U/L, TGO = 300 U/L), pentru care este transferat la o altă clinică pediatrică din spital. Se exclud la acel moment, dintre cauzele infecțioase ale citolizei, infecția cu virusurile hepatitice, B, C, VEB, toxoplasmoza. Se remarcă însă serologie pozitivă pentru infecția cu CMV - fracția IgM (rezultat survenit după externare).
Urmează un interval de 5 ani și jumătate (4 luni → 5 ani și 11 luni) în care pacientul nu a mai avut nici o internare în Spitalul „Sfânta Maria” (mama declară că o perioadă de timp au locuit într-o altă țară - Italia). Din biletele de externare (Azienda Ospedaliera Universitaria Integrata VERONA - prof. Dott. Maria Carli) constatăm că la 5 ani se adresează acestui spital din Italia prezentând clinic tumefacție pulsatilă evidentă clinic parietal drept. Efectuează RMN și CT craniocerebral, cu evidențierea unei lacune osoase parieto-occipitale pentru care se intervine neurochirurgical și se efectuează cranioplastie. Prezența strabismului convergent de la nivelul ochiului stâng este corelată la acel moment cu TCC și i se recomandă corecție optică.
La 5 ani și 11 luni, copilul revine în țară și un nou control neurochirurgical relevă o citoliză hepatică importantă de 20xN (TGP = 637 U/L, TGO = 274U/L), iar CT-ul craniocerebral de control descrie atrofie corticală sechelară parietală dreaptă, plastie cu material osos, minimă deviație de sept nazal și hipertrofie adenoidiană. În Clinica II, pacientul ajunge în luna mai a anului curent, la vârsta de 6 ani și două luni, prin transfer de la Spitalul de Boli Infecțioase, unde a fost evaluat pentru citoliză, fără a se evidenția o cauză infecțioasă.
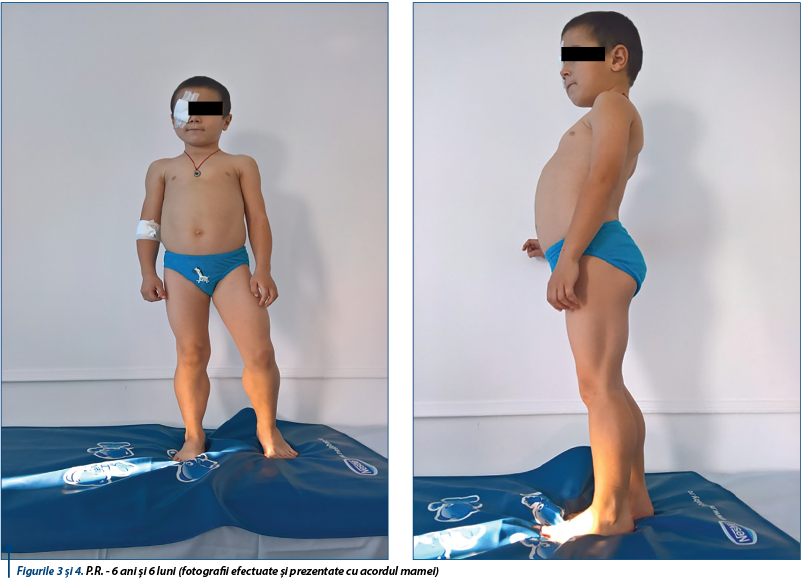
La examenul clinic se constată morfometrie normală, alopecie liniară cicatriceală postoperatorie regiunea parietală dreaptă; pseudohipertrofie musculară (gambe și coapse bilateral), ce îi imprimă pacientului un aspect atletic; la palpare, această musculatură are o fermitate excesivă, se remarcă contractura tendonului lui Achile, copilul acuză oboseală musculară la mers, la urcatul scărilor, la alergat - acțiuni pe care le efectuează cu greutate, manevra GOWERS este pozitivă, asimetrie sternală, stern bombat (aspect în carenă) la nivelul hemitoracelui stâng, hiperlordoză lombară, mers și alergat pe vârfuri, pulmonar - SaO2(-) = 99%, suflu sistolic gradul II/6 parasternal stâng, AV și TA în limite normale, abdomen proeminent din cauza hiperlordozei, ROT prezente bilateral, simetrice, strabism congenital de ochi stâng (figurile 3, 4, 5).
Anamneza cu citoliză hepatică persistentă din antecedente și examenul clinic cu modificările musculare menționate au ridicat suspiciunea unei miopatii. Din acest motiv s-au determinat enzimele musculare și s-a evidențiat faptul că citoliza era de cauză extrahepatică, o miocitoliză (CPK-9000 U/L, LDH = 1478U/L, TGO = 169 U/L, TGP = 346 U/L). Au urmat încadrarea miopatiei în una din bolile musculare și excluderea altor miopatii.
Am exclus miopatiile inflamatorii, care sunt rare la copil (anamnestic, clinic și biologic), dintre miopatiile metabolice am luat în discuție boala Pompe (glicogenoza tip II a) din cauza CPK-ului crescut și a prezenței hipotoniei, dar a fost exclusă pe baza absenței hepatosplenomegaliei, a suferinței cardiace de tip cardiomiopatie dilatativă, pe baza testului pentru boli lizozomale negativ și a examenului histopatologic ulterior. Distrofiile miotonice - boala Steinert - a fost exclusă pe baza aspectului clinic (care este tipic în această afecțiune), absenței afectării oculare de tip cataractă, absenței endocrinopatiilor prezente în această afecțiune, pe valorile CPK-ului (normale sau ușor crescute) și examenul histopatologic. Miotonia congenitală - forma AR: Becker a fost exclusă prin severitate și debut mai tardiv, iar forma AD: Thomsen prin tabloul clinic și evoluție. Paramiotonia congenitală Eulenburg a fost exclusă prin faptul că are debut în perioada de sugar, afectează musculatura pleoapelor, a feței, a faringelui. Distrofiile musculare congenitale sunt rare și se caracterizează prin hipotonie de la naștere, apariția contracturilor înainte de 6 luni, enzimele musculare fiind normale sau ușor crescute. Miopatiile congenitale, diferite de distrofiile congenitale, au, de asemenea, debut neonatal, cu diminuarea progresivă a ROT, iar enzimele musculare sunt normale; diagnosticul stabilindu-se prin biopsie musculară.
Aspectul clinic a pledat pentru o distrofie musculară progresivă, condiționată genetic. Distrofiile musculare AD și AR au tablou clinic diferit de cel al cazului nostru. Dintre DMP X-linkate am exclus distrofia musculară Becker - debut mai tardiv al simptomelor (nu înainte de adolescență, de obicei în jurul vârstei de 12 ani, iar 90% din cazuri înainte de 20 ani), evoluție mai lentă a afectării musculare față de DMD, pierderea mersului în decada a patra de viață, la unii cardiomiopatia domină tabloul clinic, precum și prin examenul histopatologic.
Biopsia musculară a fost edificatoare pentru diagnosticul de distrofie musculară de tip Duchenne: mușchi striat în care se remarcă inegalitatea diametrelor fibrelor musculare striate, arii întinse de fibre musculare necrozate, alături de fibre musculare regenerate, precum și fibre musculare omogenizate, ușoară fibroză la nivelul endomisiului și a perimisiumului, prezența de arii de țesut adipos alb.
Examenul genetic a inclus test ADN (MLPA) la copil și la mamă, determinarea CPK-ului la mamă (cu valori crescute: 353U/L, N = 24-170), realizarea arborelui genealogic, din care remarcăm faptul că nici în familia tatălui, nici în familia foarte numeroasă a mamei (10 frați) nu a fost nici o persoană cu afectare musculară (figura 6).
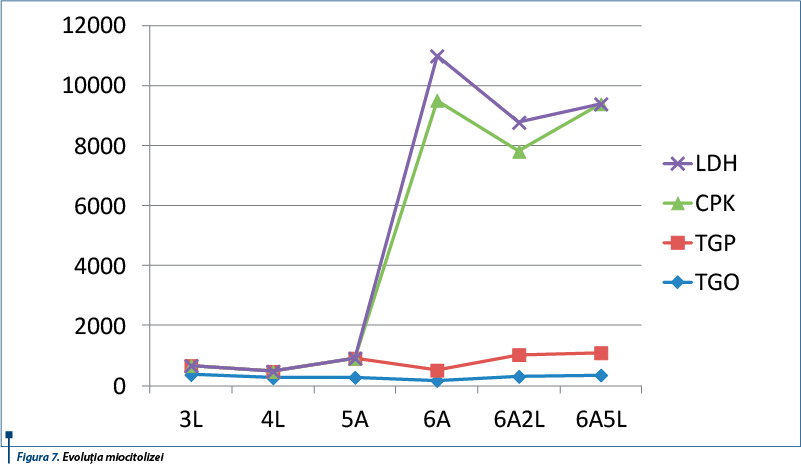
Tabloul biologic actual a evidențiat o hemoleucogramă normală (GA = 6.260/mm3, cu formulă leucocitară echilibrată – PMN = 40,3%, Ly = 49,7%, Mo = 6,7%, Eoz = 3%, GR = 4,96 mil./mm3, Ht = 40,1%, Hb = 13,2 g/dl, Tr = 338.000/mm3), absența inflamației, miocitoliză importantă (TGO = 176 U/L, TGP = 358 U/L, CPK = 8.966 U/L, LDH = 1.622 U/L) (figura 7), glicemie, funcție renală, metabolism fosfocalcic în limite normale.
Paraclinic: EKG - traseu cu aritmie sinusală 62-92/min, A QRS = +50 grade, PQ = 0,12 sec., suprasolicitare ventriculară stângă, posibil biventriculară, schițează bloc minor de ram drept, fără modificări de repolarizare ventriculară, QT = 0,35 sec. (limita superioară).
Diagnosticul pozitiv: distrofie musculară progresivă Duchenne, infecție congenitală cu CMV, strabism convergent OS, bloc minor de ram drept.
Tratamentul este simptomatic și de susținere; necesită implicare multidisciplinară - pediatrică, neuropsihiatrică, psihologică, oftalmologică, cardiologică, pneumologică, ortopedică, genetică.
Obiective:
- Prevenirea contracturilor musculare, menținerea tonusului muscular și a funcției musculare și implicit a deformărilor osteoarticulare, pentru prelungirea cât mai mult a momentului dependenței de cărucior.
- Menținerea funcției pulmonare.
Kinetoterapie susținută, orteză gleznă-picior pentru prevenirea contracturii tendonului lui Achile; alungirea chirurgicală a tendonului în caz de apariție a contracturii. Fizioterapia este necesară și pentru menținerea funcției respiratorii. Se recomandă activități ușoare, înotul; sedentarismul/statul la pat agravează funcția musculară. Se utilizează corset pentru trunchi în vederea prevenirii apariției scoliozei, bretele toracice speciale, intervenții chirurgicale de fixare a coloanei. Când bolnavul devine dependent de cărucior, necesită supraveghere permanentă.
Evoluție și complicații. Evoluția bolii este severă, cu apariția deformărilor articulare prin contracturile musculare și ale tendoanelor, scolioză, hiperlordoză; în stadiile tardive - imposibilitatea mersului, ce determină deplasare în scaun cu rotile (în jurul vârstei de 9-10 ani); dacă evoluția este mai lentă, această dependență apare în jur de 12-13 ani.
Complicațiile care pot apărea în evoluție sunt pulmonare, ca urmare a deformărilor cutiei toracice, ale coloanei vertebrale, a hipotoniei musculare (infecții pulmonare severe, hipoventilație nocturnă, insuficiență respiratorie cronică restrictivă). Alte complicații care pot apărea - afectare cardiacă, neuropsihică. De asemenea, terapia cortizonică susținută are numeroase efecte secundare bine cunoscute, miopatia cortizonică fiind dificil de diferențiat de afectarea musculară din cadrul bolii.
Prognosticul bolii este sever, decesul survine în adolescență, până la vârsta de 25 de ani (prin infecții respiratorii severe, insuficiență respiratorie cronică, insuficiență cardiacă refractară la tratament).
Particularitatea cazului constă în întârzierea diagnosticului, prin necomplianța familiei; concomitența la un pacient de 6 ani a unei DMD cu o infecție congenitală cu CMV, ambele afecțiuni asociind citoliză și afectare oculară.
Cercetări
Corticosteroizii (prednisolon, deflazacort) determină, pentru scurt timp (până la 2 ani), îmbunătățirea rezistenței și funcției musculare(3). Mecanismul prin care corticoterapia este eficientă în DMD este insuficient cunoscut, dar se pare că este legat de modularea apoptozei, inflamației, reglarea concentrației de calciu și de miogeneză.
De asemenea, s-a demonstrat că terapia cortizonică zilnică, instituită precoce, determină creșterea forței musculare, prelungirea duratei de mers, deși unele studii arată că argumentele în acest sens nu sunt foarte puternice(4).
Corticoterapia pe termen lung întârzie pierderea mersului cu câțiva ani, reduce necesitatea intervențiilor chirurgicale la nivelul coloanei vertebrale, îmbunătățesc funcția cardio-respiratorie și, în general, îmbunătățesc calitatea vieții(5).
Se recomandă inițierea corticoterapiei înainte de 4-6 ani; nu se recomandă înainte de vârsta de 2 ani. Întârzierea introducerii corticoterapiei după pierderea capacității de mers se pare că determină prezervarea forței musculare la nivelul membrelor superioare, scade progresia scoliozei și întârzie declinul funcției cardio-respiratorii(6).
Cea mai nouă terapie în DMD, în prezent, este exon skipping. Mecanismul ce stă la baza exon skipping este reprezentat de o mutație specifică antisense oligonucleotide (AON). O oligonucleotidă antisens este un polimer sintetic scurt, de acid nucleic, format din maximum 50 de perechi de baze, care se leagă la nivelul exonului ce conține mutația, astfel încât, când gena este translată de la nivelul mARN, aceasta este „sărită”, obținându-se o proteină funcțională(7). Există două tipuri de oligo-antisens:
- 2’-O-methyl phosphorothioate oligos (Drisapersen).
- Morpholino oligos (Eteplirsen) - testate în trialuri clinice.
Francesco Muntoni, un renumit profesor neurolog pediatric la Imperial College London și care în prezent își desfășoară activitatea la UCL Institute of Child Health Londra, în cadrul Departamentului de cercetare a bolilor neuromusculare, și-a dedicat întreaga carieră cercetării aspectelor clinice, patologice și genetice în DMD. Și-a adus o contribuție importantă în cercetare prin descoperirea faptului că afectarea cardiomiopatică dilatativă X-linkată este secundară anomaliei genei distrofinei. O altă contribuție pe care a adus-o în cadrul bolilor neuromusculare a fost descoperirea a doi loci noi și a 3 gene implicate în distrofiile musculare congenitale.
În 2011, prof. Muntoni și echipa sa au publicat în Lancet un studiu cu privire la exon skipping și „repararea” distrofinei la pacienții cu DMD, după administrarea sistemică a Phosphorodiamidate morpholino oligomer (un studiu open-label de fază 2), în doze progresiv crescătoare. Cei 19 pacienți incluși în studiu au avut vârste cuprinse între 5 și 15 ani. Li s-a efectuat biopsie musculară înainte de inițierea tratamentului intravenos cu AVI-4658 și după 12 săptămâni de tratament. Ei au demonstrat, pe de o parte, sigurața și tolerabilitatea acestui medicament, iar pe de altă parte, au evidențiat prin RT-PCR, imunohistochimie și imunoblottting faptul că acest medicament are capacitatea de a induce exon 51 skipping și repararea distrofinei(8,9).
În 2012, Franceso Muntoni a publicat un alt articol, în Molecular Therapy, în care demonstrează, de data aceasta, că expresia distrofinei este însoțită de creșterea expresiei a-sarcoglycan, b-dystroglycan și neuronal nitric oxide synthase (nNOS) la nivelul sarcolemei, fiecare din acestea fiind o componentă a unui subcomplex diferit al distrofinei asociate cu complexul glicoproteic(10). Expresia nNOS a fost relocată în sarcolema pacienților cu DMD, la care deleția distrofinei a lăsat intact domeniul ce leagă nNOS (exonii 42-45). Rezultatele au demonstrat că distrofina indusă prin exon 51 la pacienții cu deleție poate restaura complexul asociat distrofinei și poate rămâne funcțională. Întrucât DMB prezintă o variantă mai ușoară a mutației distrofinei, în care pacienții își pierd mult mai târziu capacitatea de a merge, s-a încercat transformarea mutației DMD în mutația DMB, tot prin metoda exon skipping, la nivelul mARN-ului(11).
Acest lucru a fost demonstrat in vivo pe modele animale(12). Dintre cele două clase de medicamente utilizate in vivo, 2¢O-methyl-ribooligonucleoside-phoshophorothioate (2¢OMe) și phosphorodiamidate morpholino oligomers (PMOs) s-a demonstrat că ultimul este mai eficient(13).
Tratamentul cu celule stem constă în înlocuirea celulelor musculare cu celule stem. Acestea pot fi obținute prin două metode:
- transferul de celule stem care implică celule de la pacientul cu DMD, ce sunt modificate genetic in vitro pentru restabilirea expresiei distrofinei și care sunt apoi reimplantate(14);
- a doua metodă: transfer allogenic de celule stem de la un individ cu distrofină funcțională la un pacient cu DMD(15).
Deși celulele stem izolate din mușchi au abilitatea de a se diferenția în miotubuli când sunt injectate în mușchiul animalelor, acestea își pierd abilitatea de a se răspândi sistemic. Pentru a furniza o doză terapeutică unui singur mușchi, acesta ar trebui să fie injectat cu celule stem la fiecare 2 mm. Această problemă a fost evitată prin utilizarea altor celule stem multipotente, numite PERICYTE, care sunt localizate în interiorul vaselor de sânge din mușchi.
După ce trec prin vase, acestea au capacitatea de a fuziona și de a forma miotubuli; prin urmare, pot fi injectate intraarterial, trec de peretele arterial și ajung în mușchi, unde se diferențiază în fibră musculară potențial funcțională. Aceste celule sunt extrase, crescute în culturi, apoi injectate în fluxul sanguin, de unde există posibilitatea ca ele să ajungă în mușchiul scheletic afectat. Terapia cu celule stem poate îmbunătăți regenerarea musculară și repararea distrofinei(16).
În 2007, cercetătorii au realizat, pentru prima oară, terapie genică mediată viral. Biostrophin este un vector de eliberare utilizat în terapia genică a DMD. S-au mai utilizat adenovirusuri și/sau retrovirusuri ca vectori cu abilitate de a transmite mesajul genetic pentru distrofină în fibra musculară în care aceasta lipsește.
În martie 2016, în Cell Research, un grup de autori din SUA publică date despre o nouă terapie promițătoare în DMD: CRISPR/Cas9. Este o nouă metodă de editare a unei noi gene pentru a corecta mutația ce conduce la DMD (model pe șoarece). CRISPR/Cas9 livrat prin anevo-virusuri poate elimina cu precizie mutația din gena distrofinei, permițând mecanismelor de reparare a ADN-ului să o înlocuiască cu o copie normală a genei. Beneficiul acestei tehnici de terapie genică este că poate corecta permanent defectul dintr-o genă decât să adauge tranzitor una funcțională(17,18,19,20). Acest procedeu nu este încă fezabil la om.
În 2012, un grup de cercetători de la Institutul de Boli Cardiovasculare din Cedars - Sinai, a publicat în Science un studiu randomizat, dublu orb controlat cu placebo, de fază 3, în care au demonstrat eficiența tadalafilului în îmbunătățirea fluxului sangvin în mușchiul striat al pacienților cu DMD și DMB, prin ocolirea „defectului” și stimularea căii NO-cGMP. Tadalafilul previne oboseala musculară post-efort și spasmul de la nivelul vaselor de sânge(21). De asemenea, simvastatinul și-a demonstrat efectele remarcabile pe care le are în creșterea forței musculare la șoareci.
În 2015 au fost publicate în Lancet rezultatele unui studiu dublu orb controlat cu placebo, de fază 3, prin care s-a dovedit eficacitatea tratamentului cu idebenone (raxone) - derivat sintetic al CoQ10 la pacienții cu DMD. Se arată că acest medicament reduce semnificativ declinul anual al PEF, cu 66% față de placebo; de asemenea, și alți parametri, precum FVC și FEV1, sunt ameliorați(22).
Prevenirea bolii (a nașterii unui copil cu DMD) se face prin sfat genetic ce se recomandă tuturor femeilor care au deja un băiat afectat de această boală sau o rudă afectată; este necesară identificarea femeilor purtătoare, prin examinarea pedigree-ului acestora și determinarea CPK-ului (crescut în >75% din cazuri) și, nu în ultimul rând, diagnosticul prenatal prin amniocenteză.
Bibliografie
1. Marzese DM, Mampel A, Gomez LC, Echeverria MI, et al. Detection of deletions and duplications in the Duchenne muscular dystrophy gene by the molecular method MLPA in the first Argentine affected families. In Genet. Mol. Res. 2008; 7 (1): 223-233.
2. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010; 9: 77-93.
3. Biggar WD, Harris VA, Eliasoph L, et al. Neuromusc Disord 2006; 16:249-255.
4. Bushby K, Muntoni F, Hughes R, Griggs R. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids, Naarden, The Netherlands. Neuromuscul Disord 2004; 14: 526-534.
5. Shapiro F, Zurakowski D, Bui T, Darras B.T. Progression of spinal deformity in wheelchair-dependent patients with Duchenne muscular dystrophy who are not treated with steroids: coronal plane (scoliosis) and sagittal plane (kyphosis, lordosis) deformity. Bone Joint J 2014; 96: 100-105.
6. Moxley R.T. 3rd, Pandya S, Ciafaloni E, Fox D.J, Campbell K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol 2010; 25: 1116-1129.
7. Harding, PL, Fall AM, Honeyman K, Fletcher S, Wilton SD. The influence of antisense oligonucleotide length on dystrophin exon skipping. Molecular. Therapy 2007; 15 (1): 157-66.
8. Kinali M, Arechavala-Gomeza V, Feng L. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009; 8:918-928.
9. Nguyen TM, Morris GE. Use of epitope libraries to identify exon-specific monoclonal antibodies for characterization of altered dystrophins in muscular dystrophy. Am J Hum Genet. 1993; 52:1057-1066).
10. Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C, Morgan J, Muntoni F. Restoration of the Dystrophin-associated Glycoprotein Complex After Exon Skipping Therapy in Duchenne Muscular Dystrophy. Molecular Therapy 2012; 20 2, 462-467.
11. Arechavala-Gomeza V, Graham IR, Popplewell LJ, Adams AM, Aartsma-Rus A, Kinali M et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum Gene Ther 2007; 18: 798-810.
12. Yokota, T, Lu, QL, Partridge, T, Kobayashi, M, Nakamura, A, Takeda, S et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 2009; 65: 667-676.
13. Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol 2009; 8: 918-928.
14. Mendell J, Clark K. Challenges for gene therapy for muscular dystrophy. Curr Neurol Neurosci Rep 2006; 6: 47-56.
15. Partridge T. Stem cell therapies for neuromuscular diseases. Acta Neurol Belg 2004; 104: 141-147.
16. Markert C, Atala A, Jennifer K, Cann BA, Christ G, Furth M, Ambrosio F, Martin K. Mesenchymal Stem Cells: Emerging Therapy for Duchenne Muscular Dystrophy. PM R. 2009. 1(6): 547-559.
17. Long C, Amoasil L, Mireault AA, et al., Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy, Science 2016; 351:400-403.
18. Nelson CE, Hakim CH, Ousterout DG, et al., In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy, Science. 2016; 351:403-407.
19. Tabebordbar M, Zhu K, Cheng JK, et al., In vivo gene editing in dystrophic mouse muscle and muscle stem cells, Science 2016; 351:407-411.
20. McNally EM, Kaltman JR, Benson DW, et al., Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy, Circulation 2015; 131:1590-1598.
21. Martin EA, Barresi R, Byrne BJ et.al. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy and in boys with Duchenne muscular dystrophy. Science, 2012.
22. Buyse GM, Voit T, Schara U et al, Efficacy of Ibedenone on respiratory function in pactients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet, 2015.

Sindromul de compartiment după mușcătura de șarpe
Cristina Jităreanu, Irina-Mihaela Ciomagă, Elena Tarcă, Sidonia Susanu, Nicolai Nistor
În lume, numărul anual de mușcături de șerpi este estimat la peste 5 milioane, determinând aproximativ 130.000 de decese/an, în special în regiunea tropicală. Regiunile cele mai afectate sunt Africa Subsahariană și Asia ...
Evaluarea nutriţională cu particularităţi evolutive la pacienţii cu mucoviscidoză
Evelina Moraru, Ramona Diaconu, Aurica Rugină
Mucoviscidoza este cea mai frecventă maladie monogenică, autozomal recesivă, în populațiile de origine caucaziană, cu evoluție cronică, letală, cu o incidență de 1/2.500 de nou-născuți (o frecvență a heterozigoților de 1...
Despicăturile labio-maxilo-palatine
Elena Ţarcă, Bogdan A. Stana, Simona Gavrilescu, Bogdan Savu
Despicăturile labio-maxilo-palatine sunt cele mai frecvente malformaţii congenitale ale regiunii cefalice şi implică o diversitate de anomalii la nivelul buzei superioare şi maxilarului superior, uneori şi a nasului şi a aparatului auditiv, implicând o lipsă de substanţă uni- sau bilaterală la nivelul zonei a...
Evaluarea nutriţională cu particularităţi evolutive la pacienţii cu mucoviscidoză
Evelina Moraru, Ramona Diaconu, Aurica Rugină
Mucoviscidoza este cea mai frecventă maladie monogenică, autozomal recesivă, în populațiile de origine caucaziană, cu evoluție cronică, letală, cu o incidență de 1/2.500 de nou-născuți (o frecvență a heterozigoților de 1...
Despicăturile labio-maxilo-palatine
Elena Ţarcă, Bogdan A. Stana, Simona Gavrilescu, Bogdan Savu
Despicăturile labio-maxilo-palatine sunt cele mai frecvente malformaţii congenitale ale regiunii cefalice şi implică o diversitate de anomalii la nivelul buzei superioare şi maxilarului superior, uneori şi a nasului şi a aparatului auditiv, implicând o lipsă de substanţă uni- sau bilaterală la nivelul zonei a...