Rolul imunoglobulinei E în patologia pediatrică
Role of Immunoglobulin E in Pediatric Pathology
Abstract
IgE implications in pathology, although vaguely known and accepted, only recently have been unified and described in terms of complex immunological mechanisms. Classically considered as a marker of allergy/atopy, IgE hides many other facets through involvement in infectious and parasitic diseases. Hyper-IgE syndrome is now explained by genetic mechanisms and it presents through a variety of clinical phenotypes. Cytokine interactions and immunological pathogenetic implications are still to be deciphered. In contrast, hypo-IgE syndrome is an emerging pathogenic entity recently described. Pathologies related to IgE capture and captivate through complexity. Its understanding leads to new ways of therapy.Keywords
immunoglobulins EpathogenesischildrenRezumat
Implicaţiile IgE în patologie, deşi vag cunoscute şi acceptate, abia de curând au fost unificate şi descrise din prisma mecanismelor imunologice complexe. Considerată clasic ca marker al alergiei/atopiei, IgE ascunde multe alte valenţe prin implicarea în patologii infecţioase, pulmonare, parazitare. Sindromul hiper-IgE este în prezent explicat prin genetica elucidată, se prezintă clinic printr-o varietate de fenotipuri, iar imunologic interacţiunile citokinice şi implicările patogenice sunt încă în încercări de a fi descifrate. La polul opus, sindromul hipo-IgE se conturează ca entitate patogenică foarte recent descrisă, fiind în lumina cercetărilor actuale. Patologia legată de IgE surprinde şi captivează prin complexitate, iar toate cuceririle fiziopatogenice deschid căi importante spre terapie.Cuvinte Cheie
imunoglobuline EpatogeniecopilImunoglobulinele E: rol, sinteză, nivel seric
În 1968, două grupuri de cercetători, din SUA şi Suedia, au anunţat descoperirea unei noi imunoglobuline, care a fost denumită IgE. La persoane predispuse genetic (atopici - prezintă unul sau mai multe teste cutanate pozitive la alergenii comuni din mediu) se produce o secreţie crescută a acestei clase particulare de imunoglobuline. IgE este prezentă şi în serul persoanelor sănătoase, dar în cantitate foarte mică. IgE se produce faţă de o varietate mare de antigene numite alergeni. Alergenii nu produc nici un fel de reacţie la o persoană sănătoasă, în schimb pot determina boli alergice la persoanele atopice prin reacţii de hipersensibilizare de tip I, respectiv prin modificările produse de imunoglobulinele E (reagine).
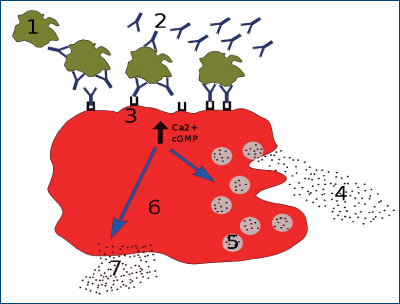
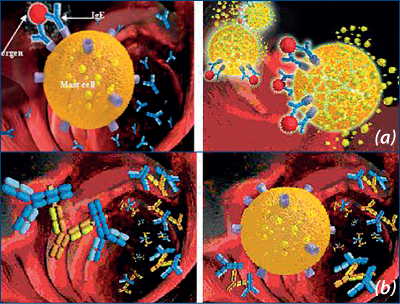
Imunoglobulinele E se găsesc în principal în secreţiile mucoase de la nivelul tractului gastrointestinal şi respirator, în ser fiind prezente în concentraţii foarte mici. Aceste niveluri circulante foarte scăzute se datorează afinităţii mari a mastocitelor pentru IgE, de care se leagă prin receptorul Fce. Rata de sinteză este, de asemenea, foarte scăzută, IgE ataşându-se de mastocite şi bazofile şi, astfel, activând eozinofilele. IgE pe suprafaţa mastocitelor au un timp de înjumătăţire de peste 10 zile. Spre deosebire de alte imunoglobuline care se leagă de receptorii Fc doar când antigenul e legat de un anticorp, IgE se leagă de FceR şi în absenţa antigenului, „sensibilizând” mastocitul să se degranuleze când antigene multivalente se leagă încrucişat de FceR (figura 1).
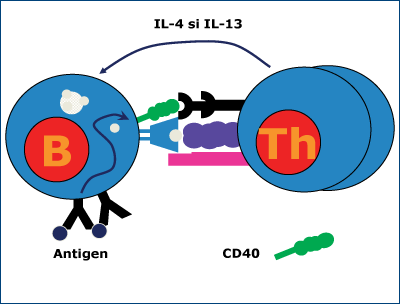
Formarea IgE se realizează în mai multe etape: 1) antigenul întâlneşte celula B; 2) celula B îmbracă antigenul şi îl prezintă celulei T helper; 3) celula B proliferează şi 4) switch-ul spre IgE cu participarea celulelor plasmatice şi a celulelor de memorie.
Acest switch către IgE este un proces cu trei niveluri de semnalizare:
1. Prezenţa antigenului care controlează întregul proces, activând celula B (figura 2);
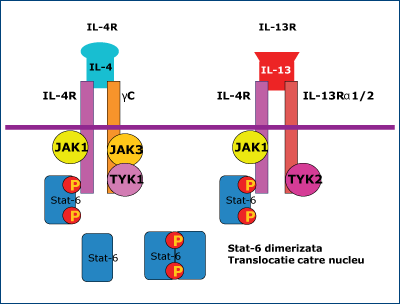
2. Activarea celulei B indusă de antigen, care duce la inducerea STAT6 prin intermediul IL-4 şi IL-13 (figura 3);
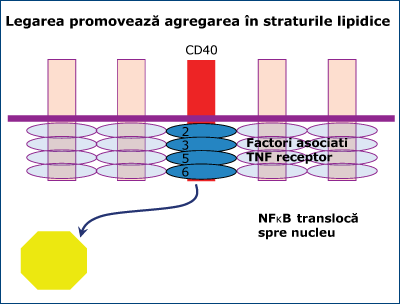
3. Eliberarea NFkB din IkB prin intermediul CD40L din celulele T helper (figura 4).
Semnificaţii patologice ale nivelului seric
de IgE
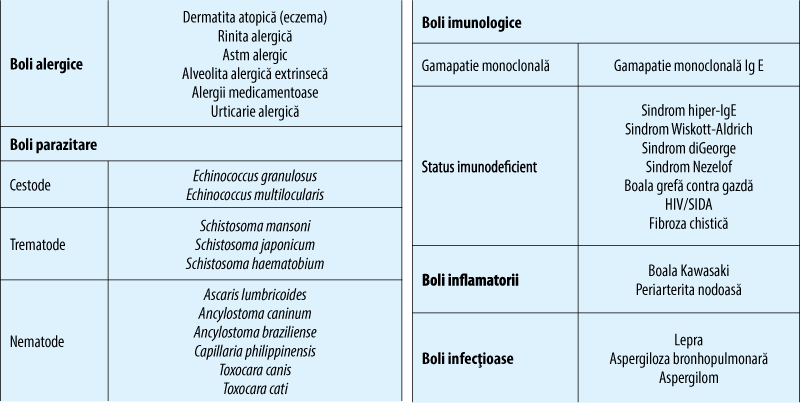
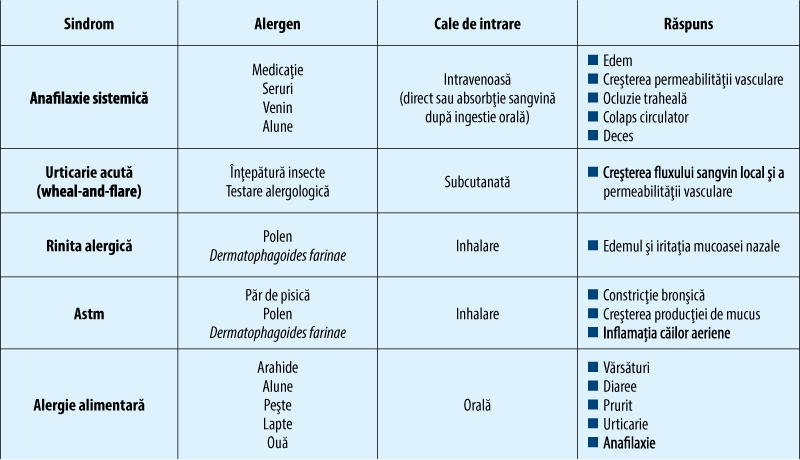
Creşteri ale nivelului IgE sunt înregistrate în afecţiuni alergice, boli parazitare, tulburări imunologice, boli infecţioase, boala Hodgkin, mielom multiplu de tip IgE, cele mai frecvente dintre acestea fiind redate în tabelul 1. Manifestările clinice majore ale afecţiunilor alergice IgE mediate sunt anafilaxia, astmul, rinita alergică, dermatita atopică, alergia alimentară, alergia la înţepături de insecte, alergia la lapte.
Cuantificarea IgE serice în practica clinică este întotdeauna indicată în prezenţa anumitor afecţiuni cum ar fi: gamapatia monoclonală IgE, sindromul hiper-IgE, aspergiloza bronhopulmonară alergică sau sindroamele de imunodeficienţă. Uneori, indicaţia cuantificării IgE serice este necesară la pacienţi cu suspiciune de boală alergică sau boală parazitară helmintică. Nu există indicaţie de cuantificare IgE la pacienţi cu suspiciune de boală inflamatorie sau infecţii neparazitare.
La copii, creşteri ale IgE peste 20 U/mL susţin diagnosticul de manifestare alergică; totuşi, o valoare normală nu exclude o boală alergică. La adulţi, IgE sunt mai puţin folositoare în stabilirea etiologiei alergice a simptomelor:
-
IgE >1 DS peste medie sugerează boala alergică;
-
IgE >2 DS peste medie susţin puternic etiologia alergică.
Cuantificarea nivelului IgE nu primează dacă etiologia alergică a fost probată clinic, iar valori crescute ale IgE au valoare limitată în predicţia tendinţei alergice. Reacţiile alergice IgE-mediate se regăsesc în multe patologii inflamatorii şi infecţioase (tabelul 2).
Sindromul hiper-IgE
Sindromul de hiper-IgE (HIES) este o imunodeficienţă primară complexă, caracterizată printr-un spectru de anomalii care interesează sistemul imun, oasele, ţesutul conjunctiv şi dinţii.
Boala mai este cunoscută ca sindromul JOB (sindromul IOV), din cauza aspectului „fiert” al pielii, o marcă a sindromului, ţinând cont şi de reminiscenţa referitoare la personajul biblic Iov, care a fost lovit de Satana „cu o fierbere de la creştetul capului până la tălpile picioarelor”(1). Cu acest citat din „Cartea lui Iov” - Davis, Schaller şi Wedgewood au inventat în 1966 conceptul de „sindromul lui Iov”(12). În plus, Buckley (sindromul Buckley, 1973) este considerat că a descris pentru prima dată sindromul clasic hiper-IgE.
HIES include imunodeficienţe primare rare cauzate de triada clinică:
-
niveluri serice crescute de IgE (>2.000 UI/ml);
-
abcese stafilococice recurente ale pielii;
-
pneumonie caracterizată prin complicaţia cu pneumatocele.
Nivelurile IgE serice pot varia în cadrul acestui sindrom, în funcţie de vârsta pacientului:
-
adulţi: >2.000 UI/ml - definitorii, în prezenţa fenomenelor inflamatorii sau a pneumoniei;
-
la copii, care au niveluri în mod normal scăzute, sindromul hiper-IgE se consideră la valori de 10 ori mai crescute ale IgE pentru vârstă;
-
cordon ombilical: <1 U/mL (1 U = 2,4 ng);
-
nivelurile stabile ca la adult se ating la vârsta de 5-7 ani;
-
între 10-14 ani se poate depăşi nivelul de la adult;
-
după 70 de ani scad uşor.
Principalele manifestări imunologice sunt:
-
abcese cutanate recurente;
-
leziuni cutanate asemănătoare cu dermatita atopică suprainfectată;
-
infecţii sino-pulmonare;
-
niveluri crescute IgE;
-
chemotaxie neutrofilică anormală.
Principalele manifestări non-imunologice sunt:
-
modificări cranio-faciale şi scheletice;
-
anomalii dentare;
-
osteopenie;
-
complicaţii neurologice;
-
hernii.
În determinismul genetic al HIES se citează o ştergere interstiţială de pe cromozomul 4q21. Din punct de vedere fiziopatologic au fost descrise mutaţii negative transdominante în gena ce codează un factor de transcripţie - STAT3 (transductorul semnalului şi activatorul transcripţiei 3). În 2008, un colectiv condus de Cindy Ma a arătat că posibile defecte ale Th17 datorate mutaţiilor STAT3 ar determina sindrom hiper-IgE(3).
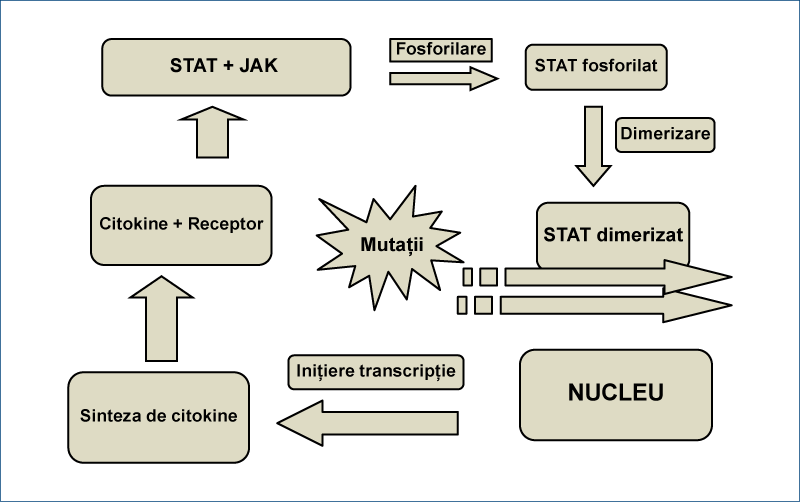
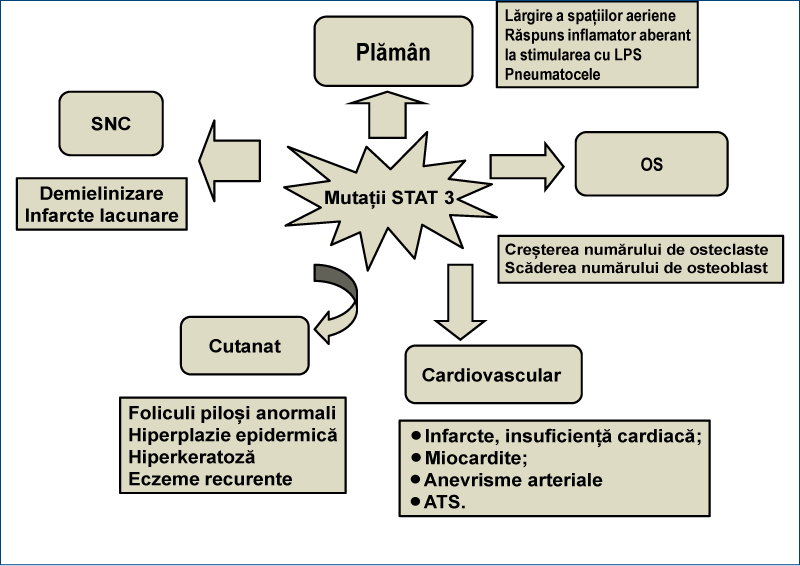
Gena STAT se află pe cromozomul 17q21 şi este activată în funcţie de nivelul citokinelor. Molecula STAT se leagă de receptorul JAK printr-un proces de fosforilare disociază şi se formează STAT fosforilat, care suferă un proces de dimerizare, rezultând STAT dimerizat. Acesta este transportat la nucleu, unde participă la iniţierea transcripţiei, stimulând astfel sinteza de citokine, cu formarea complexului citokină-receptor. Mutaţiile afectează STAT dimerizat, ciclul se întrerupe la acest nivel, în final rezultând fie o sinteză deficitară de citokine, fie sinteza normală de citokine cu funcţii modificate(2).
Aceste mutaţii reprezintă semnalul pentru receptorii citokinelor şi conduc la o generare defectuoasă a Th17, subset al celulelor T CD4+ efectorii(3). Deficienţa STAT3 scade expresia RORgt şi RORa, cunoscuţi ca factori de transcripţie specifici pentru celulele Th17, având astfel un rol esenţial în programarea expresiei celulare genetice a Th17. STAT3 este astfel deosebit de importantă în expresia IL‐21 şi diferenţierea celulară Th17 mediată de IL-21.
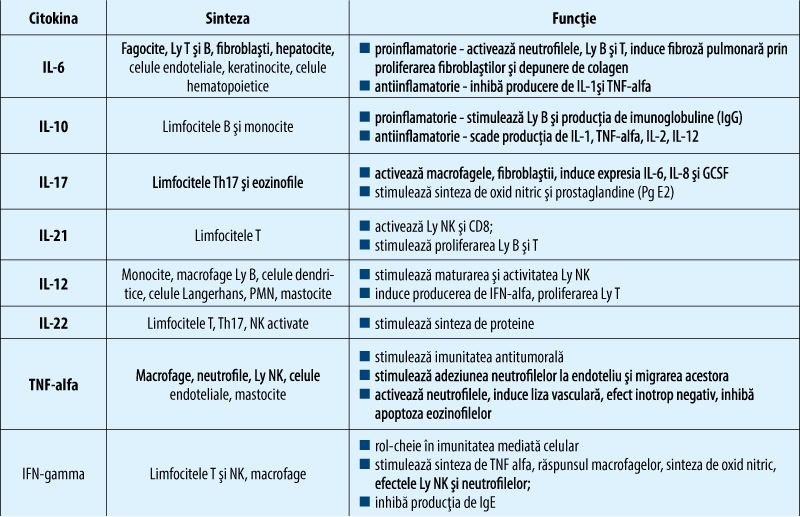
Citokinele acţionează asupra celulelor imunocompetente, fiecare având un rol deosebit în expresia clinică a fenomenelor inflamatorii; cel mai frecvent implicate citokine sunt IL-6, IL-10, IL-12, IL-17, IL-21, IL-22, TNF-alfa, IFN-gama, funcţiile acestora fiind explicate în tabelul 3.
Citokinele implicate determină imunofenotipul bolii. Astfel, IL-6, care în mod normal reglează o proteină MCP1, la pacienţii cu HIES fiind în cantităţi reduse, sunt potenţate efectele proinflamatorii. IL-17, IL-21, IL-22 au rol în apărarea împotriva fungilor şi bacteriilor extracelulare (candidozele, suprainfecţiile pulmonare, infecţiile cutanate recurente). Deficitul de IL-17 determină hipereozinofilia, iar IL-10 joacă un rol în potenţarea efectului proinflamator, hiper-IgE şi eozinofilie(2).
Expresia clinică a mutaţiilor STAT3 este schiţată în figura 6.
Manifestările cutanate în sindromul
hiper-IgE (HIES)
O erupţie în perioada de nou-născut este de obicei prima manifestare clinică a HIES. Erupţiile cutanate încep de obicei pe faţă şi scalp în primele săptămâni de viaţă şi sunt de obicei pustuloase şi eczematoide. Biopsiile arată infiltrate eozinofilice, iar în culturile de bacterii creşte de obicei Staphylococcus aureus.
Erupţii cutanate adesea persistă pe tot parcursul copilăriei, dar pot fi controlate cu terapii antistafilococice, constând în antibiotice, antiseptice de actualitate sau ambele. Aspectul de piele „fiartă”, de asemenea, începe devreme în viaţă şi este aproape o regulă în HIES, cu excepţia cazului în care tratamentul antistafilococic a fost iniţiat precoce.
Clasic, semnele inflamatorii, cum ar fi căldura şi tumefacţia dureroasă, sunt variabile, uneori duc la apariţia de abcese reci. Tratamentele corect instituite pot ameliora mult aspectul pielii, dar în zonele cu probleme, cum ar fi axila şi regiunea inghinală, aspectul „fiert” poate persista.
Anomalii musculo-scheletice
Anomaliile musculo-scheletice în HIES includ scolioza, fracturi la traumatisme minime, osteopenie, laxitatea articulaţiilor şi boli degenerative comune. Scolioza este comună şi apare adesea în timpul adolescenţei, similar scoliozei idiopatice. Fracturi la traumatisme minime apar la aproximativ jumătate din persoanele cu HIES şi implică frecvent coastele şi oasele lungi.

Osteopenia şi osteoporoza pot de asemenea să apară, dar par să fie independente de fracturi. Resorbţia osoasă mediată de osteoclaste este anormală în HIES şi probabil se referă la osteopenie şi fracturi. În ciuda acestui fapt, vindecarea osoasă după fracturi sau după intervenţiile chirurgicale este de obicei normală. Hiperextensibilitatea articulaţiilor mici şi mari este frecventă şi poate contribui la începutul unei boli degenerative comune, în special la nivelul coloanei vertebrale cervicale.
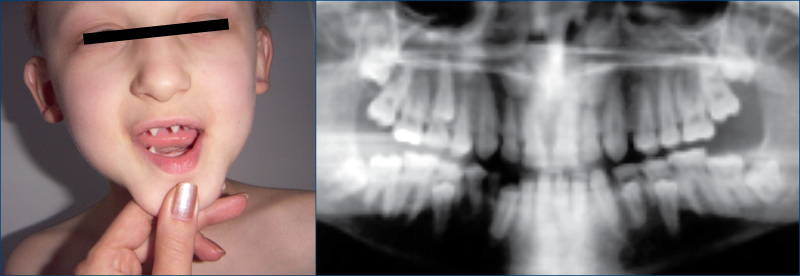
Anomaliile dentare
Majoritatea indivizilor cu HIES nu reuşesc să îşi înlăture dentiţia primară (de lapte) în mod normal, necesitând adesea extragerea chirurgicală a unora sau a tuturor dinţilor primari, pentru a permite dentiţiei secundare să se dezvolte. Variaţii caracteristice ale mucoasei bucale, limbii, cerului gurii şi obrajilor includ depresiuni centrale ale limbii care devin susceptibile la infecţii cu Candida şi deformări ale proeminenţelor centrale ale palatului (figura 8).
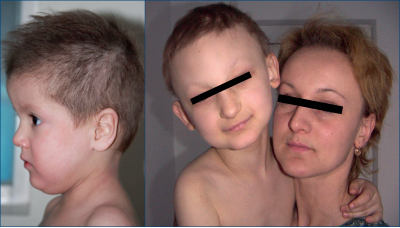
Anomalii ale capului şi feţei
Caracteristicile distinctive faciale ale pacienţilor cu sindrom hiper IgE s-au dovedit a fi generale, cu apariţie până la vârsta de 16 ani. Pacienţii prezintă asimetrie facială, cu o sugestie de hemihipertrofie facială; fruntea este proeminentă, ochii înfundaţi în orbite şi un pod nazal larg; fosele nazale sunt cărnoase şi prezintă uşor prognatism. Faţa are trăsături dure şi este aspră la palpare. Distanţa interalară a fost comparată cu valorile publicate standard şi cu scoruri Z pentru rasa albă. Circumferinţa capului este mărită, cu tendinţa de a fi mai mare decât în mod normal (figura 9).
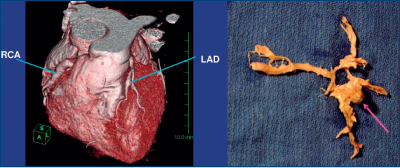
Anomalii vasculare
Anomaliile vasculare sunt recent descrise în HIES. Anevrismele coronare care determină frecvent infarcte miocardice au determinat o examinare mai atentă a anomaliilor arteriale. Tomografia computerizată şi IRM arată că dilatarea şi torsionarea arterei coronare, cu formarea de anevrisme, sunt frecvente. Au fost descrise şi infarcte lacunare (figura 10).
Afecţiuni maligne
HIES este asociat cu un risc crescut de limfom non-Hodgkin cu celule B şi histologie agresivă. Alte afecţiuni maligne raportate au inclus limfom Hodgkin, leucemii, cancer de vulvă, ficat şi plămâni.
Sindromul hiper-IgE necesită colaborare interdisciplinară prin complexitatea simptomatologiei clinice. Ponderea acestor manifestări este dictată de forma bolii, fiind cunoscute trei forme distincte ale sindromului hiper IgE:
-
forma autozomală dominantă;
-
forma autozomală recesivă (rară);
-
forma sporadică.
Principalele semne clinice întâlnite cu o frecvenţă crescută în forma autozomală dominantă şi în cea sporadică sunt sintetizate în figura 11.
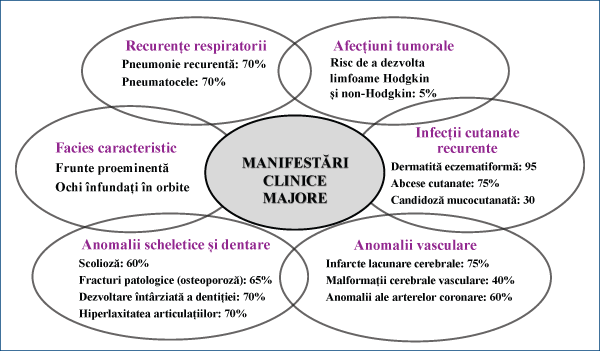
Mortalitatea în această formă de boală se corelează cu:
infecţii pulmonare sau la nivelul SNC cu Pseudomonas aeruginosa, Aspergillus fumigatus, Scedosporium prolificans;
-
hemoptizii fatale;
-
limfom Hodgkin sau non-Hodgkin.
Fenotipul non-imunologic este determinat de mutaţii STAT3 din viaţa embrionară cu prognostic letal, defecte ale funcţiei cardiomiocitelor, eozinofilie, osteopenie, formare de chisturi pulmonare. Toate grupurile rasiale sunt afectate, fără preferinţă de gen. Gena activatoare de transcriere 3 (STAT3) se află pe cromozomul 17. Majoritatea mutaţiilor HIES au sediu obligatoriu în ADN, domeniul SH2 şi domeniile de transactivare. Domeniul SH2 mediază dimerizarea STAT3. Domeniul transactivării conţine site-uri de fosforilare implicate în activarea STAT3. Mutaţiile identificate până în prezent sunt heterozigote şi se anticipează a fi proteine pozitive, datorită modificărilor încrucişate. Majoritatea mutaţiilor sunt multiple independente şi sunt situate în ADN şi domeniul SH2, cele mai comune fiind R382W/Q şi respectiv V637M/L.
În forma autozomal recesivă, care este mult mai rară, manifestările clinice sunt însă mai severe, asociate cu vasculite şi alte manifestări autoimune, fără manifestări sistemice (dermatită atopică, infecţii cutanate recurente, nu se formează pneumatocele). Se asociază cu mutaţii ale genei TYK2 situată pe braţul lung al cromozomului 4. Defecte în semnalele citokinice în răspunsul imun înnăscut şi dobândit determină un tablou clinic complex determinat de perturbarea diferenţierii Th1 şi accelerarea diferenţierii Th2, cu rol indispensabil al TYK2 în maturarea imunitară(4). Mortalitatea este mare în copilărie.
În 2004, au fost analizaţi 13 pacienţi provenind din 6 familii cu HIES, familii cu infecţii recurente (pneumonie şi abcese reci), eczeme şi IgE crescute, eozinofilie. Pacienţii au îndeplinit criteriile de diagnostic pentru HIES. Deoarece toţi pacienţii au provenit din familii consangvine, pare rezonabilă ipoteza unei transmiteri autozomale recesive (AR-HIES). La pacienţii cu AR-HIES sunt diagnosticate, în plus faţă de infecţiile bacteriene recurente (Staphylococcus aureus, Haemophilus influenzae, Proteus mirabilis, Pseudomonas aeruginosa şi Cryptococcus neoformans), şi infecţii cronice refractare cu Molluscum contagiosum, infecţii cu herpes simplex, care conduc la cheratită, sau infecţii recurente cu varicella zoster.
Patognomonice pentru AR-HIES sunt nivelurile IgE foarte crescute şi o eozinofilie excesivă. În plus, apar niveluri serice crescute ale altor izotipuri de imunoglobuline, ceea ce sugerează o stimulare generală a sistemului imun umoral. Eozinofilia în AR-HIES este mai pronunţată decât în forma tradiţională şi poate ajunge la niveluri de până la 17.500/ml (normal <700/ml). Această eozinofilie excesivă este incriminată şi în apariţia manifestărilor neurologice din AR-HIES(5).
Posibilităţi terapeutice
Nu există tratament specific pentru HIES în prezent, astfel încât tratamentul se bazează pe administrarea unei medicaţii antifungice, antibiotice şi/sau antitermice - în funcţie de simptomatologie, tratamentul chirurgical al abceselor localizate (chirurgie toracică), administrare de imunoglobuline specifice.
Din 2009 se discută posibilitatea unei noi terapii cu anticorpi monoclonali specifici - omalizumab (Merck, 2009). Acesta este un agent imunoterapeutic folosit ca anti-IgE, dar cu eficacitate terapeutică mai mult pe astmul sever, la pacienţii sub 12 ani, cu boală alergică diagnosticată şi controlată insuficient cu corticosteroizi inhalatori. Prognosticul acestor pacienţi ar putea fi mult îmbunătăţit.
O altă posibilitate terapeutică o reprezintă transplantul de măduvă, iar în viitor - posibil terapie genică, datorită descoperirii mutaţiilor STAT3.
A fost emisă şi ipoteza existenţei unei legături între mutaţia genei STAT3 şi apariţia astmului la copii, pornind de la demonstraţia faptului că mutaţii ale genei STAT3 (signal transducer and activator of transcription 3) stau la baza producerii sindromului hiper-IgE. În acest sens, au fost efectuate studii având drept obiectiv determinarea legăturii dintre mutaţiile STAT3 sau polimorfismul mononucleotidic al STAT3 (single-nucleotide polymorphisms - SNPs) şi nivelurile crescute de IgE la copiii astmatici. În urma studiilor s-a demonstrat că nu există asociere între variante STAT3 şi astm, rinită alergică sau eczemă. De asemenea, s-a demonstrat că IgE totale, IgE specifice şi numărul eozinofilelor nu s-au asociat cu modificări ale parametrilor funcţionali respiratori. Deci mutaţiile STAT3 nu influenţează IgE seric la copiii astmatici(6).
Sindromul hipo-IgE
Sindromul hipo-IgE este definit de valori ale IgE < 2 U/mL la copii şi <4 U/mL la adulţi. Deficitul de IgE este dificil de definit deoarece în mod normal nivelurile IgE sunt scăzute. Această definiţie se aplică pentru copiii sau adulţii care prezintă valori izolat scăzute pentru IgE, în timp ce valorile altor Ig (A, G, M) sunt normale.
Valori scăzute ale IgE pot fi decelate în cadrul unor afecţiuni cum ar fi: imunodeficienţa severă combinată, sindrom hiper-IgM, ataxie-telangiectazie, agamaglobulinemie Bruton, imunodeficienţă comună variabilă, hipogamaglobulinemia tranzitorie a sugarului.
Pacienţii cu deficite de IgE au prevalenţa mai mare a deficitelor imunoglobulinice (deficitul selectiv de IgA este prezent la 1,12% din 532 de pacienţi cu hipogamaglobulinemie IgE), a bolilor autoimune şi a reactivităţii non-alergice a căilor aeriene, comparativ cu pacienţii cu IgE normal(7).
Asocierea între bolile autoimune şi hipo-IgE a fost explicată prin trei mecanisme principale:
1. deficitul de IgE determină o creştere a absorbţiei antigenului la nivelul mucoasei;
2. deficitul de IgE determină o scădere a numărului, dar şi a duratei de viaţă a mastocitelor;
3. deficitul de IgE determină o scădere a ratei de producţie a anticorpilor.
IgE se găseşte predominant la nivelul mucoaselor. Astfel se explică protecţia pe care o oferă moleculele de IgE împotriva autoimunizării prin scăderea absorbţiei sistemice a antigenelor la nivelul mucoaselor, blocând principalele căi de inducere a bolii autoimune: răspunsul autoimun declanşat de limfocite prin mimetism molecular, promovarea formării de complexe imune, activarea policlonală a limfocitelor indusă de antigen şi/sau prin inducerea aberantă de antigene din clasa II MHC.
În unele studii, efectuate pe şoareci, mastocitele s-au dovedit a fi intermediari esenţiali în mecanismul de inducere a toleranţei imune mediate de limfocitele T reglatorii; este posibil, prin urmare, ca deficitul de IgE să predispună la boli autoimune prin afectarea funcţiei şi duratei de viaţă a mastocitelor.
De asemenea, este posibil ca factori genetici să predispună un individ atât la boală autoimună, cât şi la deficit de IgE, sau că nivelurile scăzute de IgE reflectă doar un dezechilibru între limfocitele Th1 şi Th2, iar înclinarea balanţei în favoarea limfocitelor Th1 ar putea media apariţia unor boli autoimune cum ar fi lupusul eritematos sistemic sau artrita reumatoidă.
În ceea ce priveşte predispoziţia la hiperreactivitate bronşică a pacienţilor cu hipo-IgE, există studii experimentale care pot oferi o explicaţie pentru apariţia inflamaţiei căilor aeriene de natură non-infecţioasă, non-alergică la unii pacienţi cu deficit IgE. Kang şi colaboratorii(8) au demonstrat apariţia bolii inflamatorii a căilor aeriene la şoarecii cu deficit în limfotoxina a (LTa-/-), asociată cu diminuarea nivelului de IgE seric şi capacitatea de reacţie redusă a căilor respiratorii după expunerea la un antigen. Boala inflamatorie pulmonară la aceşti şoareci LTa-/- este Th1-mediată şi s-a observat ameliorarea simptomelor după administrare de IgE. Autorii acestui articol sugerează că prezenţa unui nivel scăzut de IgE afectează capacitatea mastocitelor de a răspunde în mod normal la antigenele din căile respiratorii şi, în consecinţă, de a produce citokine care favorizează dezvoltarea Th2 (IL-4, IL-13).
Studii recente au arătat faptul ca deficitul de IgE predispune la boală sinopulmonară cronică cu germeni patogeni cum ar fi Streptococcus pneumoniae, Haemophilus influenzae şi Moraxella catarrhalis(9).
Secord şi colaboratorii au raportat o incidenţă mai mică a infecţiilor cu germeni oportunişti şi a malnutriţiei severe la copiii infectaţi cu HIV şi cu valori crescute ale IgE, faţă de copiii HIV pozitivi, dar cu niveluri scăzute sau normale ale IgE şi cu valori similar scăzute ale Ly T CD4+. Anticorpii anti-HIV de tip IgE au fost detectaţi la 43% din copiii cu hiper-IgE(10). Pellegrino et al. au constatat că toţi membrii unui grup de copii supravieţuitori pe termen lung, născuţi din mame HIV pozitive, aveau niveluri ridicate de IgE totale serice şi au sintetizat Ac anti-HIV-1 tip IgE capabili să inhibe replicarea HIV in vitro. Replicarea virală nu a mai fost inhibată când IgE a fost eliminat prin metode de imunoabsorbţie sau anticorpi anti-IgE(11). Într-un studiu care a implicat 700 de subiecţi asimptomatici din Tanzania, Bereczky şi colaboratorii au constatat că valori crescute ale anticorilor anti-P. falciparum de tip IgE (dar nu IgG) au fost asociate cu un risc redus pentru apariţia ulterioară a malariei evidente clinic. Există, de asemenea, studii care demonstrează că anticorpi de tip IgE pot oferi imunitate împotriva infecţiei cu B. burgdorferi şi să contribuie la eliminarea de paraziţi intestinali, cum ar fi Necator americanus(12).
Manifestările clinice ale sindromului hipo-IgE sunt nespecifice, majoritatea pacienţilor prezentând simptome respiratorii persistente care adesea sunt interpretate ca fiind de natură alergică, însă testarea cutanată este în majoritatea cazurilor negativă. Pe lângă patologia respiratorie, pacienţii mai pot asocia: artralgii, fatigabilitate şi o predispoziţie crescută a bolilor autoimune. S-a constatat, de asemenea, că aceşti pacienţi prezintă infecţii respiratorii mai severe, iar tratamentul se adresează simptomatologiei, neexistând, la acest moment, posibilitatea administrării de imunoglobuline specifice.
Bibliografie
1. Belohradsky BH, Daumling S, Kiess W, Griscelli C. The hyper-IgE syndrome (Buckley-or Job-syndrome). Ergeb Inn Med Kinderheilkd 1987;55:1-39.
2. Heimall J, Freeman A, Holland SM. Pathogenesis of hyper IgE syndrome, Clin Rev Allergy Immunol. 2010;38(1):32-8 Review.
3. Ma CS, Chew JYG, Simpson N et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008:205(7);1551-1557.
4. Minegishi Y, Saito M, Karasuyama H. The Hyper IgE Syndrome and Mutations in TYK2, Immunity 2009;26(5):526-536.
5. Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes, Immunol Rev. 2005;203:244-50.
6. Wjst M, Lichtner P, Meitinger T, Grimbacher B - STAT3 single-nucleotide polymorphisms and STAT3mutations associated with hyper-IgE syndrome are not responsible for increased serum IgE serum levels in asthma families. Eur J Hum Genet 2009;17:352-356.
7. Smith JK, Krishnaswamy GH, Dykes R, Reynolds S, Berk SL. Clinical manifestations of IgE hypogammaglobulinemia. Ann Allergy Asthma Immunol. 1997;78(3):313-8.
8. Kang HS, Blink SE, Chin RK, Lee Y, Kim O, Weinstock J, Waldschmidt T, Conrad D, Chen B, Solway J, Sperling AI, Fu YX: Lymphotoxin is required for maintaining physiological levels of serum IgE that minimizes Th1-mediated airway inflammation. J Exp Med 2003;198:1643-1652.
9. Pate MB, Smith JK, Chi DS, Krishnaswamy G. Regulation and dysregulation of immunoglobulin E: a molecular and clinical perspective. Clinical and Molecular Allergy 2010; 8:3.
10. Secord EA, Kleiner GI, Auci DL, Smith-Norowitz T, Chice S, Finkielstein A, Nowakowski M, Fikrig S, Durkin HG. IgE against HIV proteins in clinically healthy children with HIV disease. J Allergy Clin Immunol 1996;98:979-984.
11. Pellegrino MG, Bluth MH, Smith-Norowitz T, Fikrig S, Volsky DJ, Moallem H, Auci DL, Nowakowski M, Durkin HG. HIV type 1-specific IgE in serum of long-term surviving children inhibits HIV type 1 production in vitro. AIDS Res Hum Retroviruses 2002;18:363-372.
12. Pritchard DI, Walsh EA: The specificity of the human IgE response to Necator americanus. Parasite Immunol, 1995;17:605-607.

Managementul infecţiei pulmonare la pacienţii cu mucoviscidoză (fibroză chistică)
Ioana Ciucă, Zagorca Popa, Liviu Laurenţiu Pop
Mucoviscidoza (fibroza chistică - FC) este cea mai frecventă boală monogenică potenţial letală a populaţiei de origine caucaziană, cu transmitere autozomală recesivă. Deşi manifestările sunt extrem de variate, afectarea ...
Antibioterapia în pneumonia comunitară
Paraschiva Cherecheș-Panța
Pneumonia comunitară rămâne o problemă de sănătate la copil. În lumina abordărilor actuale, e necesară o apreciere a severităţii pentru măsurile iniţiale şi terapia ulterioară, fie în ambulator fie, dacă este cazul, în spital. Antibioterapia se impune de primă intenţie în cazurile selectate, cu deteriorare...
Recomandări actuale în managementul şi urmărirea astmului bronşic la copil
Bogdan A. Stana
Astmul bronşic reprezintă cea mai frecventă boală cronică a copilăriei şi principala cauză de morbiditate, având drept consecinţe absenteism şcolar, adresabilitate crescută către unităţile de primire urgenţe şi spitalizări repetate. Simptomatologia apare deseori în perioada copilăriei, mai frecvent la sexul m...
Managementul infecţiei pulmonare la pacienţii cu mucoviscidoză (fibroză chistică)
Ioana Ciucă, Zagorca Popa, Liviu Laurenţiu Pop
Mucoviscidoza (fibroza chistică - FC) este cea mai frecventă boală monogenică potenţial letală a populaţiei de origine caucaziană, cu transmitere autozomală recesivă. Deşi manifestările sunt extrem de variate, afectarea ...
Antibioterapia în pneumonia comunitară
Paraschiva Cherecheș-Panța
Pneumonia comunitară rămâne o problemă de sănătate la copil. În lumina abordărilor actuale, e necesară o apreciere a severităţii pentru măsurile iniţiale şi terapia ulterioară, fie în ambulator fie, dacă este cazul, în spital. Antibioterapia se impune de primă intenţie în cazurile selectate, cu deteriorare...