Elemente actuale privind fiziopatologia cirozei hepatice la copil
Updates regarding the pathophysiology of liver cirrhosis in children
Abstract
Liver cirrhosis in pediatric age is still a tough reality, both through the problems related to diagnosis and treatment and also by the idea of fatality, in the impossibility of liver transplantation. Although there are remarkable advances in deciphering the pathogenic mechanisms, with new encouraging therapeutic options, the difficulties in the management of the cirrhotic child remain, along with the impossibility of using all therapeutic resources at an early age. The authors make an insight into the current pathophysiology of liver cirrhosis, with its specificities at pediatric age. Childhood cirrhosis has etiological and evolutionary features related to immunological, nutritional, progression and decompensation factors.Keywords
cirrhosischildpathogenesisRezumat
Ciroza hepatică la vârsta pediatrică reprezintă încă o realitate dură, atât prin problemele de diagnostic şi tratament, cât şi prin ideea fatalităţii, în imposibilitatea transplantului hepatic. Deşi există progrese remarcabile în descifrarea mecanismelor patogenice, cu deschiderea unor porţi terapeutice încurajatoare, dificultăţile în managementul copilului cirotic rămân cele de ordin material, alături de imposibilitatea utilizării unor resurse terapeutice la vârsta mică. Autorii realizează o incursiune în fiziopatogenia actuală a cirozei hepatice, cu particularităţile acesteia specifice vârstei pediatrice. Copilul nu trebuie privit ca un adult în miniatură; ciroza copilului are particularităţi etiologice şi evolutive, legate de aspectele imunologice, nutriţionale, de progresie şi de decompensare.Cuvinte Cheie
cirozăcopilpatogenieDefinition and generalities
Liver cirrhosis is a serious symptomatic complex, caused by progressive and generally irreversible liver damage, which causes the destruction of the lobular architecture by extensive fibrosis, nodular regeneration, and intermittent necroinflammatory processes(1).
Liver cirrhosis is the final stage of all chronic liver diseases. In fact, cirrhosis can overlap with the underlying liver disease, making it difficult to determine the nature of the initial injury(2).
Cirrhosis and its complications are health problems that mainly affect adults and are less common in children, unless they occur in association with innate metabolic abnormalities or developmental disorders. There are two chronic liver pathologies in children, less known and not found in adulthood, which have a cirrhogenic evolution at pediatric age:
a) A non-Wilsonian form of cirrhosis with hepatic overload of copper and generally fatal outcome, which was endemic in India, known as Indian children’s cirrhosis (ICC). It is found under different names, but it has often liver histological changes identical to those of cirrhosis. The etiology of this disease remained elusive and debated, discussing excessive copper intake through the use of copper tools or household water provided through copper pipes. However, a large multicenter study conducted a few years ago in India disproved this hypothesis and suggested the possibility of a transient genetically determined abnormality in copper metabolism during childhood, which makes the liver susceptible to an unknown toxic agent(3).
The natural history of the disease has changed in the last two decades and the detection of ICC in the active progressive phase seems to have suddenly decreased over time.
b) Another cause of cirrhosis at pediatric age in the current global wave is related to non-alcoholic fatty liver disease (NAFLD), associated with obesity and various components of metabolic syndrome, which affects not only the adult population, but also children and adolescents(4,5).
In the United States of America, NAFLD is currently reported to be the most common cause of chronic liver disease in the pediatric population, leading to marked liver fibrosis and even cirrhosis in this age group(6).
There is a wide clinical variability between the various forms of cirrhosis. In 1977, the World Health Organization defined cirrhosis as a diffuse hepatic process characterized by fibrosis and the transformation of normal hepatic architecture into abnormal structural nodules.
Cirrhosis is a dynamic state, reflecting the competition between the processes of cell damage (necrosis), the response to aggression (fibrosis) and regeneration (nodule formation). Regional liver fibrosis or the formation of isolated nodules are not necessarily cirrhosis(7).
As the cirrhosis progresses, the destruction of the hepatic architecture and the compression of the vascular and biliary structures are achieved. These essential architectural changes lead to disturbance of the supply of nutrients, oxygen and transport of metabolites in various areas of the liver and can perpetuate the cirrhotic process even though the initial trigger is controlled or eliminated(7).
Classification
Numerous classification criteria have been proposed: macroscopic and microscopic appearance of the liver, etiology of cirrhosis, clinical elements. Because cirrhosis is, in later stages, a self-sustaining process, the macro- and microscopic aspects only occasionally reveal the nature of the triggering process(8,9).
1. Histological classification
Periportal (biliary) cirrhosis is characterized by biliary stasis, generalized reduction of the bile ducts, and replacement of the liver parenchyma with connective tissue in the framework and derived from the portal tracts. The lobular structure is generally preserved.
This type of cirrhosis is common in children with biliary atresia, cystic fibrosis and progressive familial intrahepatic cholestasis (PFIC) type I. Biliary cirrhosis is accompanied by severe cholestasis, including severe jaundice, disabling pruritus and hyperchromic urine and stools(10).
Postnecrotic (irregular) cirrhosis is the result of chronic and/or recurrent destruction of hepatocytes. It is characterized by piecemeal necrosis (“piece-by-piece necrosis”) that occurs at the interface between hepatocytes and portal tracts or fibrous septa. Bridging fibrosis develops later, along with the destruction of the lobular architecture and the appearance of regenerative nodules. Thus, micronodular cirrhosis is achieved.
In children, postnecrotic cirrhosis occurs as a sequela of neonatal hepatitis. It is associated with chronic active hepatitis, caused by HBV or HCV, or as a result of autoimmune or idiopathic inflammation. Medications such as methyldopa or isoniazide, which can cause chronic active hepatitis, can lead to post-necrotic cirrhosis(11).

Cardiac cirrhosis develops as a result of centrolobular hemorrhagic necrosis. Increased pressure in the right atrium (due to congestive heart failure, congenital heart disease or constrictive pericarditis) leads to increased pressure in the hepatic vein and congestion of blood flow in the centrolobular areas. Necrosis leads to the formation of fibrous bridges between the central veins. Venoocclusive disease and Budd-Chiari syndrome, which are based on congenital or acquired obstruction of the hepatic veins, also lead to cardiac cirrhosis.
The natural history of many liver diseases eventually leads to cirrhosis, each with a histological pattern or unique clinical features. The cirrhosis in Wilson’s disease and hemochromatosis is characterized by the presence of pigments (“pigment cirrhosis”), along with the presence of regenerative nodules. Diastase-resistant intracellular inclusions observed in PAS (periodic acid-Schiff) stain sustain the a1-antitrypsin deficiency or hepatic syphilis(12).
The histological classification of cirrhosis is often of limited clinical utility, as there are many cases of cirrhosis that do not fit into a specific pattern. Many forms of liver disease have a specific histological appearance at the onset of the disease, but as cirrhosis progresses from the early to late stages, these patterns intertwine, making these classifications less useful in practice.
Anthony (1977) proposed a classification of cirrhosis into three entities, according to etiology:
-
Cirrhosis with established etiological associations (hepatitis, metabolic diseases, biliary diseases, venous obstruction).
-
Cirrhosis with questionable etiology (autoimmunity, mycotoxins, schistosomiasis, malnutrition).
-
Cirrhosis of unspecified (cryptogenic) etiology.
The grouping of diseases that progress to cirrhosis after their cause is of clinical use, as it provides a common framework for the clinical investigation of cirrhosis, for prognostic elements and genetic counseling. Because there are several similar morphological and histological patterns that occur in many diseases, this gnoseological classification has proven to be the most useful(13).
2. The main etiological categories are:
1. Postnecrotic cirrhosis
2. Biliary cirrhosis
3. Metabolic and genetic cirrhosis
4. Vascular cirrhosis
5. Nutritional cirrhosis
6. Idiopathic (cryptogenic) cirrhosis.
A) Postnecrotic cirrhosis is the most common and complicates the hepatocellular lesions caused by hepatitis viruses (HBV, HDV, HCV etc.), neonatal or toxic-drug hepatitis.
The installation interval is variable, depending on a multitude of factors (the etiological agents and regarding the host); thus, HDV aggravates and accelerates the evolution of an HBV infection, and HCV infection produces, in a larger but more frequent range, cirrhotic evolution(14). Autoimmune hepatitis progresses to cirrhosis depending on the type, age of onset and early treatment.
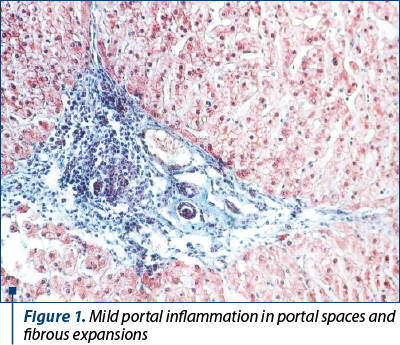
B) Biliary cirrhosis appears as a consequence of chronic cholestatic syndrome, mainly of extrahepatic or intrahepatic biliary atresia, syndromatic ductular hypoplasia (Alagille syndrome) and nonsyndromatic ones, bile ducts’ cysts and pseudocysts, recurrent familial cholestasis etc. In children, an extremely rare entity is primary biliary cirrhosis (by inhibition of lymphocytes and antimitochondrial antibodies).
Metabolic abnormalities of biliary acids’ synthesis, by toxic effects of aberrant products, can cause biliary cirrhosis(12).
C) Metabolic and genetic cirrhosis occur through:
-
abnormalities of carbohydrate metabolism – congenital galactosemia, hereditary intolerance to fructose and glycogenosis types III (Fabry Forbes) and IV (Andersen);
-
abnormalities of lipid metabolism (Niemann-Pick, Gaucher, Wolman diseases, Bassen-Komzweig disease or a-b-lipoproteinemia);
-
mucopolysaccharidosis (Hurler and Hunter diseases);
-
metabolic abnormalities of copper (Wilson’s disease), iron (primary or secondary hemochromatosis), coproporphyrins (hepatic porphyria).
Other genetic causes, such as cystic fibrosis, coprostanic acidemia or Byler’s disease, can also progress to cirrhosis.
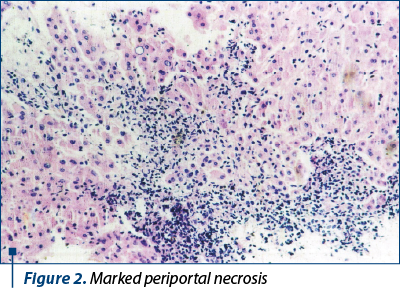
D) Vascular cirrhosis leads to portal hypertension due to prehepatic, intrahepatic or posthepatic obstructions. This mechanism is found in portal cavernoma, portal vein thrombosis/stenosis or congenital portal system compression. In evolution, other diseases can lead to circulatory involvement, with serious consequences: congenital hepatic fibrosis, veno-occlusive disease, pericarditis(8).
E) Nutritional cirrhosis occurs as a result of the prolonged nutritional impairment and severe (malnutrition), or due to the effect of the alcohol in utero or after birth (fetal alcohol syndrome), toxic alkaloid food (teas) and after severe poisoning(15).
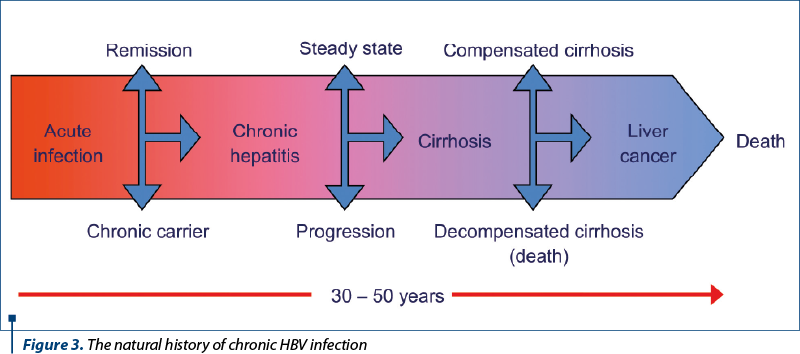
3. The clinical classification of cirrhosis depends on the competitive processes of hepatocyte destruction, fibrosis and regeneration. If cell necrosis exceeds the capacity of the liver to regenerate, then signs and symptoms of hepatocellular failure occur. The inability of the liver to synthesize serum proteins and coagulation cofactors results in ascites and coagulopathies. The inability of the liver to produce or secrete bile results in the appearance of cholestasis, clinically manifested as jaundice, pruritus, hyperchromic urine and alcoholic stools. The accumulation of neurotoxic substances leads to hepatic encephalopathy.
Chronic active and neonatal hepatitis are two examples of hepatopathies associated with primary hepatocellular insufficiency. Primary biliary pathology manifests as severe cholestasis that can progress to biliary cirrhosis. It is seen in infants with extrahepatic bile duct atresia or in older children with cystic fibrosis. If fibrosis and regeneration overcome necrosis, portal hypertension appears, accompanied by specific symptoms. Hypersplenism may indicate the presence of portal hypertension, although life-threatening bleeding of esophageal varices may be the first clinical sign of presentation to the physician.
Often, the progression of cirrhosis is silent, in which case the term compensated cirrhosis is used. The patient is apparently healthy and there are no symptoms or signs of liver disease. A detailed anamnesis does not offer suggestive elements about an underlying pathology. The clinical examination may reveal hepatomegaly or splenomegaly, but there are also cases in which they are absent. The biochemical tests show moderate increases in transaminases or alkaline phosphatase levels. Decompensated cirrhosis is found in many patients during investigations, unrelated to other pathologies or as a result of family screening(16).
Liver metabolic diseases (a1-antitrypsin deficiency, Wilson’s disease) have a long compensated phase, in which hepatopathy is not suspected. There are also many cases of compensated cryptogenetic cirrhosis.
Pathophysiology
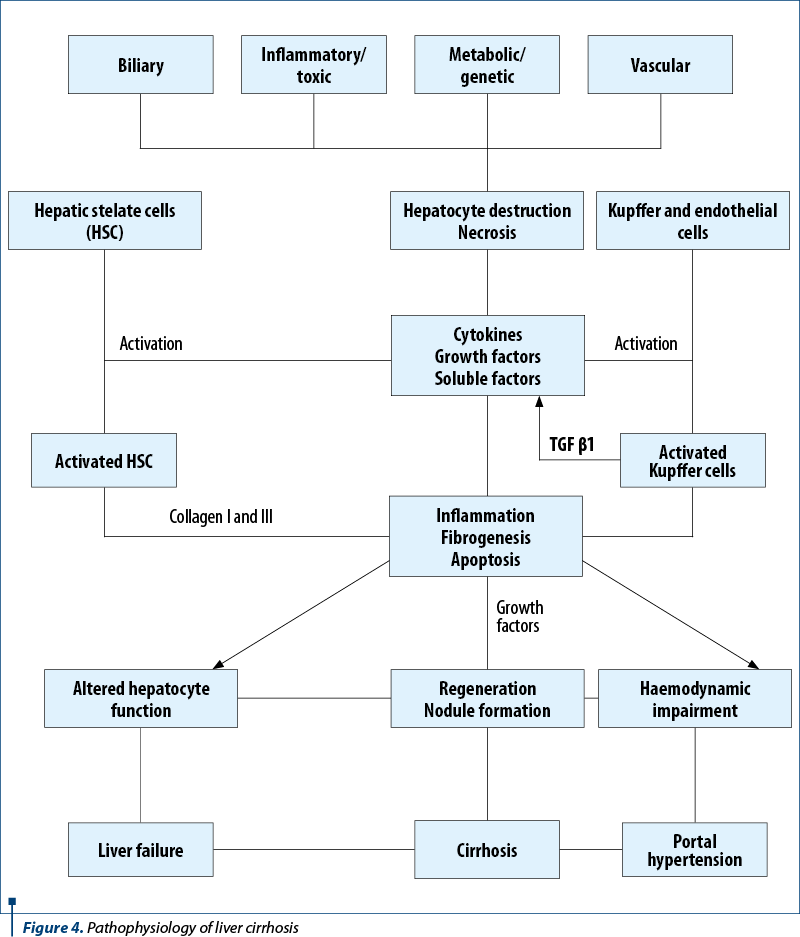
New pathophysiological hypotheses have been in focus recently regarding the pathogenesis of portal hypertension and progression to liver cirrhosis. The process itself is particularly complex, the molecular mechanisms are barely deciphered, and the involvement is systemic.
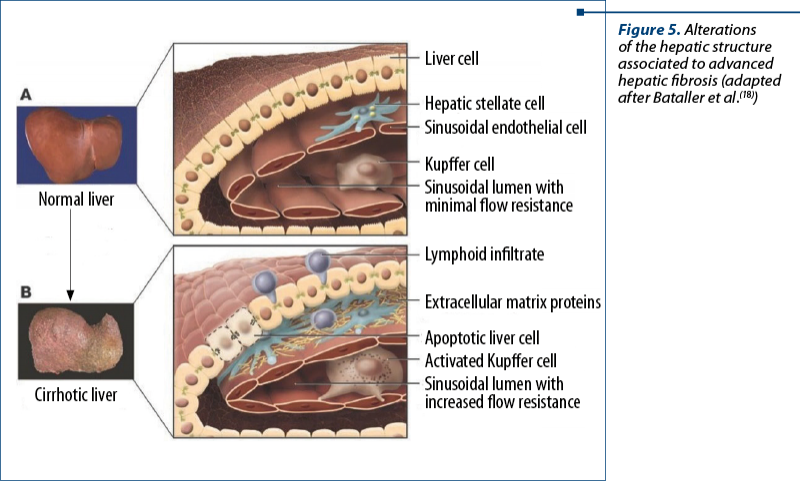
At the hepatic level, the main events governing the evolution of cirrhosis are the remodeling of the extracellular matrix, fibrogenesis and the appearance of regenerative nodules.
1. Extracellular matrix (ECM)
The first step in the process of fibrogenesis is the direct injury of the hepatocyte – the result of any type of aggression: viral, ischemic, exposure to toxins. After aggression, the parenchymal cells regenerate and replace the necrotic cells. This process is associated with inflammation and deposition of collagen-containing extracellular matrix (ECM). The cells responsible for intrahepatic fibrosis have not been fully identified. A central role in this process is played by the stellate liver cell(17).
Recent hypotheses involve the “non-resident” stem cells in the pathogenesis of cirrhosis. It is unclear whether “resident” (hepatic) or “non-resident” (extrahepatic) stem cells become or transform into non-parenchymal cells (endothelial, stellate, Kupffer) responsible for the fibrotic process. If stimulated by inflammatory cells or cytokines, hepatocytes and support cells secrete an altered ECM. ECM is essential for the proper survival and functioning of hepatocytes and provides a stable environment with tissue compartments.
ECM macromolecules can be divided into three distinct categories: collagen, proteoglycans and glycoproteins.
Collagen is the most abundant protein in ECM. There are 13 distinct types of collagen that have been described. Types I, III, IV, V and VI were isolated from human liver tissue.
Proteoglycans are associated with the basement membrane and appear to play a central role in regulating cell permeability and cell proliferation. Proteoglycans are composed of a protein covalently linked to at least one glycosaminoglycan. In the liver, heparan sulfate is the most abundant glycosaminoglycan.
Glycoproteins make the connection between the extracellular matrix and the surrounding cells. Fibronectin and laminin are the most abundant glycoproteins in the liver, both of which are multifunctional matrix proteins with multiple domains. Fibronectin (with at least 10 isoforms) mediates cell adhesion to collagen, increases granulation tissue, and is a chemoattractant and growth factor for mesenchymal cells. Laminin, a more studied protein, also has domains that include collagen receptors and epithelial cells. Laminin acts on hepatocytes, regulating their growth and differentiation; it also plays a role in organizing liver architecture.
In cirrhosis, ECM is altered quantitatively and qualitatively. In the normal liver, connective tissue proteins are found along the basement membrane, surrounding blood and lymph vessels, and around the bile ducts. There is also collagen in the perisinusoidal space. Hepatocytes and most portal spaces are devoid of connective tissue. In cirrhosis, type III and type IV collagen are increased in the perisinusoidal space. The sinusoids are lined with a new basement membrane, consisting of laminin and type III collagen. In addition, types IV, V and VI collagen increase 8-10 times, while laminin increases three times(19).
2. Fibrogenesis
Although initially considered permanent, there are current data suggesting that fibrogenesis is reversible, at least in some cases. Experimentally induced fibrosis in animals has recently been shown to be reversible. The reversal of fibrosis has also been shown in some liver diseases, such as chronic HCV infection and non-alcoholic steatohepatitis (NASH).
Quantifying fibrosis has been an ongoing challenge for clinicians. The gold standard of these tests was liver biopsy, associated with the fibrosis scoring system. This system includes the histological activity index (HAI): Knodell score, with Ishak modification and Metavir score. The HAI system quantifies inflammatory activity from 0 to 18, assessing periportal necrosis/inflammation, lobular necrosis and inflammation, and portal inflammation.
Biochemical tests have assessed enzymes and metabolites associated with fibrogenesis. These may be useful in monitoring the progression of fibrosis.
The amino-terminal peptide end of procollagen type III (serum procollagen peptide III – P3P) is the most studied metabolite. As type III collagen is incorporated into the extracellular matrix, P3P is excised from the type III procollagen and can be quantified in serum. P3P is elevated in acute liver diseases and positively correlates with the elevated levels of transaminases in liver necrosis. Elevated levels are also found in patients with chronic liver disease, which correlate with the degree of fibrosis(7).
Serum laminin levels are elevated in patients with active fibrogenesis, and some studies have associated them with the degree of portal hypertension. Other macromolecules associated with fibrogenesis have been found in increased amounts in patients with liver fibrosis: lysyl oxidase, prolyl-4-hydroxylase, hyaluronate, type IV collagen, 7S collagen.
FibroScan is also in clinical use, its advantage being that is a noninvasive imaging method also approved in children.
3. Regeneration
In the mature human liver, hepatocytes divide at a slow rate – only 1 in 10,000 to 20,000 hepatocytes divides at a time. If stimulated, hepatocytes may have an increased rate of proliferation. In response to viral aggression, cirrhosis, ischemia, trauma or partial hepatectomy, hepatocyte proliferation increases to replace the lost cells.
Hepatic regeneration is a complex, highly regulated process at the cellular level by autocrine and paracrine mechanisms, through cytokines, controlled themselves by prostaglandins or other cytokines(7).
Conclusions
Understanding the processes leading to the cirrhotic evolution of chronic liver diseases is the starting point for the appropriate therapeutic conduct. In the era of HBV vaccination and spectacular interferon-free therapies for viral hepatitis C, there are still chronic liver diseases with more or less rapid progression to cirrhosis. Proper etiological, clinical and histological classifications are absolutely necessary to monitor and, as far as possible, to prevent complications of cirrhosis in pediatric age.
Bibliografie
-
Bozomitu L. Ciroză hepatică şi alte cauze de hipertensiune portală la copil. Editura Fundaţiei Academice Axis, Iaşi, 2009.
-
D’Amico G, Pagliaro L. The clinical course of portal hypertension in liver cirrhosis. In: Rossi P. Diagnostic imaging and imaging guided therapy. Berlin: Springer-Verlag, 2000, 15–24.
-
Nayak NC, Chitale AR. Indian childhood cirrhosis (ICC) & ICC-like diseases: the changing scenario of facts versus notions. Indian J Med Res. 2013;137(6):1029e1042.
-
Bozic MA, Subbarao G, Molleston JP. Pediatric nonalcoholic fatty liver disease. Nutr Clin Pract. 2013;28(4):448e458.
-
Tandon N, Garg MK, Singh Y, Marwaha RK. Prevalence of metabolic syndrome among urban Indian adolescents and its relation with insulin resistance (HOMA-IR). J Pediat Endocrinol Metabol. 2013;26(11-12):1123-30.
-
Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr. 2013;162(3):496e500.e1.
-
Fernandez M, Bosch J, Garcia-Pagan JC. Current concepts on the pathophysiology of portal hypertension. Ann Hepatol. 2007; 6(1):28-36.
-
de Franchis R, Primignani M. Natural history of portal hypertension in patients with cirrhosis. Clin Liver Dis. 2001;5:645-663.
-
Abraldes JG, Bosch J. Clinical Features and Natural History of Variceal Hemmorrhage. In: Sanyal AJ, Shah V (eds.). Portal Hypertension: Pathobiology, Evaluation and Treatment, Totowa: Humana Press, 2005;167-182.
-
Hupertz V, Winans C. Portal Hypertension. In: Wyllie R, Hyams J. Pediatric Gastrointestinal and Liver Disease: Pathophysiology/ Diagnosis, 3rd edition, Elsevier Inc., 2006;950-965.
-
Vajro P, Hadchouel P, Hadchouel M, et al. Incidence of cirrhosis in children with chronic hepatitis. J Pediatr. 1990;117(3):392-396.
-
Shepherd R. Complications and Management of Chronic Liver Disease. In: Kelly D. Diseases of the Liver and Biliary System in Children. Blackwell Publishing, 2008;351-377.
-
Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 1977;55(4):521-40.
-
Sebastiani G, Vario A, Guido M, et al. Sequential algorithms combining non-invasive markers and biopsy for the assessment of liver fibrosis in chronic hepatitis. B World J Gastroenterol. 2007;13(4):525-531.
-
Miu N. Patologia hepatică a copilului. Editura Dacia, Cluj-Napoca, 1992.
-
NIEC, et al. Natural history of portal hypertensive gastropathy in patients with liver cirrhosis. Gastroenterology. 2000;119:181–187.
-
Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–930.
-
Bataller R, Brenner D. Liver fibrosis. J Clin Invest. 2005;115:209–218.
-
Ross M, Pawlina W. Histology: A Text and Atlas with Correlated Cell and Molecular Biology, 5th ed. Philadelphia :Lippincott Williams & Wilkins, 2006.

Intoleranţa ereditară la fructoză: capcană de diagnostic la sugar?
Alina Grama, Claudia Sîrbe, Alexandra Mariş, Bogdan Bulata, Mariela Militaru, Tudor Lucian Pop
Hereditary fructose intolerance (HFI) – also called fructose-1-phosphate aldolase deficiency – is a rare inherited metabolic disorder characterized by the body’s inability to metabolize fructose or its precursors (sugar,...
Sindromul de regresie caudală – afecţiune congenitală rară cu implicaţii sistemice multiple
Ioana Oprescu, Cristina Becheanu, Ruxandra Darie, Mirela Pavelescu
Caudal regression syndrome (CRS) – also known as caudal dysplasia, sacral agenesis or sacral defect with anterior meningocele – is a rare congenital condition of unknown cause which consists in a developmental defect of ...
Studiu prospectiv privind managementul durerii acute la nou-născutul cu afecţiuni chirurgicale prin corelaţia scalei de durere CRIES cu protocolul terapeutic etapizat
Valentin Munteanu, Ioana Alecsandra Munteanu, Iulia Ciongradi, Ioan Sârbu, Elena Hanganu, Bogdan A. Stana, Maria Stamatin
The last 2-3 decades have brought major changes in the management of pain in the newborn....
Modificările ghidurilor în gastroenterologia şi endoscopia digestivă pediatrică în contextul pandemiei cu SARS-CoV-2
Andreea- Mădălina Nichita, Gabriela Păduraru, Lorenza Donea, Silvia Strat, Oana Maria Roşu, Smaranda Diaconescu
he infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes the coronavirus disease 2019 (COVID-19)....
Prevenția consumului de substanțe interzise începe acasă – educația timpurie
Ileana Ioniuc, Cătălina Scorța, Bogdan A. Stana
Prevenția consumului de substanțe interzise la copii și adolescenți începe în familie, prin educație timpurie adaptată vârstei. ...