Intoleranţa ereditară la fructoză: capcană de diagnostic la sugar?
Hereditary fructose intolerance: a diagnostic trap in infants?
Abstract
Introduction. Hereditary fructose intolerance (HFI) is an autosomal recessive disease characterized by the deficiency of aldolase B activity in the liver, kidney and intestine, leading to growth failure, chronic liver or kidney disease. Case report. We present the case of a 6-month-old male with HFI who presented two episodes of acute liver failure (ALF). At the first admission in our hospital, at the age of 7 weeks, the laboratory parameters infirmed an inborn error of metabolism (IEM), the liver injury being correlated with a severe systemic infection with Pseudomonas aeruginosa, with a favorable evolution. Four months later, with the onset of complementary feeding with fruits or juices, the infant presented ALF again. HFI was confirmed by genetic tests, homozygote for A175D mutation in the aldolase B gene. After following a specific diet that consisted in the excluding of fructose, sucrose and sorbitol, the clinical features and the laboratory parameters improved considerably. Conclusions. HFI can be an extremely severe condition, even life-threatening, but with an excellent evolution when the diagnosis is quick and the diet is started as early as possible. This requires a high index of suspicion from the clinician regarding the possibility of an IEM.Keywords
hereditary fructose intoleranceacute liver failuresepsisaldolase BfructosesucrosesorbitolRezumat
Introducere. Intoleranţa ereditară la fructoză este o boală autozomal recesivă, care apare secundar deficitului de aldolază B la nivel hepatic, renal şi intestinal, determinând astfel falimentul creşterii, insuficienţă hepatică acută (IHA) sau boală cronică de rinichi/ficat. Prezentare de caz. Prezentăm cazul unui sugar în vârstă de 6 luni, de sex masculin, diagnosticat cu intoleranţă ereditară la fructoză, după două episoade de IHA. La prima internare, la vârsta de 7 săptămâni, investigaţiile de laborator au infirmat o posibilă boală de metabolism, copilul asociind o infecţie sistemică determinată de Pseudomonas aeruginosa, ceea ce ne-a făcut să interpretăm injuria hepatică în acel context, având în vedere evoluţia favorabilă după introducerea antibioterapiei. La vârsta de 6 luni, odată cu începerea diversificării şi introducerea sucurilor şi pireurilor de fructe, sugarul prezintă un nou tablou sever de IHA cu coagulopatie severă, astfel încât s-a suspectat intoleranţa ereditară la fructoză. Diagnosticul a fost confirmat de testele genetice, pacientul fiind homozigot pentru mutaţia A175D. Odată cu începerea dietei specifice de excludere a alimentelor care conţin fructoză, sucroză sau sorbitol, parametrii clinici şi biologici s-au ameliorat considerabil. Concluzii. Intoleranţa ereditară la fructoză poate fi asociată cu o evoluţie severă, fiind uneori chiar ameninţătoare de viaţă, însă un diagnostic stabilit rapid, precum şi începerea cât mai precoce a dietei specifice pot îmbunătăţi semnificativ prognosticul acestor cazuri.Cuvinte Cheie
intoleranţă ereditară la fructozăinsuficienţă hepatică acutăsepsisaldolază BfructozăsorbitolIntroduction
Hereditary fructose intolerance (HFI) – also called fructose-1-phosphate aldolase deficiency – is a rare inherited metabolic disorder characterized by the body’s inability to metabolize fructose or its precursors (sugar, sorbitol). The estimated prevalence of HFI in Europe is between 1:18.000 and 1:31.000(1). HFI was described for the first time in 1956 by Chambers and Fratt in a patient who developed severe gastrointestinal symptoms after ingesting fructose and sucrose(2). HFI typically first manifests when fructose- and sucrose-containing foods are introduced in infants, often with diversification. If large quantities of fructose are ingested, the infant develops lethargy, seizures, bloating, nausea, vomiting, abdominal pain, diarrhea, and/or progressive coma. Untreated, HFI may determine acute liver failure (ALF), renal failure and death. Children with HFI can experience a normal quality of life and life expectancy if the disease is early detected and the dietary restrictions of fructose, sucrose and sorbitol are set up quickly before permanent organ injury occurs(1).
Case report
We present the case of an infant with HFI, who presented two severe episodes of ALF (one at the age of 7 weeks old and the other at 6 months old). The infant was born at 39 weeks of gestation, by vaginal delivery. The pregnancy was normal, and he has been periodically monitored in the local hospital. There was no consanguinity of the parents and the family had no history of inherited diseases. At birth, the weight was 3500 g, the height was 51 cm, and the Apgar score was 10. He presented jaundice on the second day of life for which he received two phototherapy sessions. He started breastfeeding on his first day of life. At the age of 7 weeks, he became lethargic, presenting hematemesis and melena; all these symptoms appeared after he received for five days an acetaminophen syrup for an upper respiratory infection. He was initially admitted to the local hospital, presenting jaundice, hepatosplenomegaly, severe coagulopathy, hypoglycemia, anemia and thrombocytopenia. Due to the severe evolution with aggravating liver disease (INR 3.6, not corrected with vitamin K), after few days he was transferred to our hospital with signs of encephalopathy (second-degree coma), jaundice, hepatosplenomegaly (liver at 4 cm, spleen at 3 cm below costal margin), ascites, petechiae and bleeding at the sites of venous puncture. The initial laboratory parameters in our unit revealed increased transaminases (AST 221 U/L, ALT 158 U/L) and bilirubin levels (total bilirubin 7.19 mg/dl, conjugated bilirubin 5.65 mg/dl), hypoalbuminemia (2.6 g/dl), hypoglycemia (25 mg/dl), prolonged prothrombin time (43 s) with INR 4.8 and decreased cloathing factors (Factor V 33%, Factor VII 14%). Also, he presented moderate anemia (hemoglobin 6.9 g/dl) with negative Coombs test, leucocytosis (30.600/mm³ with neutrophilia 92%) and thrombocytopenia (67.000/mm³). Astrup parameters revealed metabolic acidosis with hyperlactacidemia (5 mg/dl). The acute phase reactants were increased, and the blood culture was positive for Pseudomonas aeruginosa. We defined the case as an ALF due to sepsis with Pseudomonas aeruginosa and possibly inborn error of metabolism (IEM). He received antibiotic treatment (meropenem associated with vancomycin), albumin infusion and erythrocytes transfusion (due to severe anemia). Because of the high suspicion of an IEM, the diet was immediately changed from breastfeeding to exclusive parenteral nutrition with glucose infusion and then enteral nutrition with soy milk. However, all investigations did not support the diagnosis of an IEM (GALT enzyme, plasmatic and urinary amino acids, urinary organic acids, acylcarnitine profile and urine nuclear magnetic resonance spectroscopy were normal), so gradually the patient returned to breastfeeding. Three weeks later, the clinical features and the laboratory parameters improved considerably, therefore he was discharged, being periodically monitored in our hospital.
During the follow-up, liver parameters were constantly within normal limits, but the infant presented repeated urinary tract infections, hypercalcemia (total calcium between 12 and 13 mg/dl), hypophosphatemia (2.2-3.1 mg/dL), low parathyroid hormone and hypercalciuria. Renal ultrasonography detected nephrocalcinosis and enlarged kidneys (Figure 1). To lower calcium levels, at the age of 6 months, we recommended the reduction of dairy product consumption and the introduction of other complementary foods (fruits, vegetables).
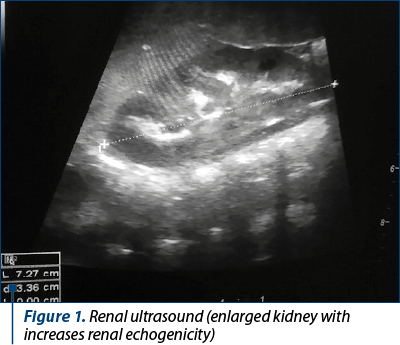
At the age of 6 months, after starting the complementary feeding with fruits and juices, the child presented in emergency with vomiting, losing weight in association with altered neurologic status, and ALF with severe coagulopathy (unmeasurable prothrombin time or INR). We performed genetic tests for aldolase B mutations (homozygote A175D), confirming the final diagnosis of hereditary fructose intolerance. After following the specific diet, the clinical symptoms disappeared and then the boy reached the normal weight and neurologic development for his age, with normal liver tests, but with the persistence of nephrocalcinosis and urinary tract infections which necessitated antibioprophylaxis with trimethoprim-sulfamethoxazole.
Discussion
We are presenting an infant who developed two severe episodes of ALF (at the ages of 2 and 6 months old). Initially, he was admitted with a highly suggestive picture for an IEM, a suspicion that was ruled out after the investigations. The diagnosis of HFI was established only at the second hospitalization when the infant developed a new episode of ALF at the onset of diversification and the introduction of fruits in the diet. Looking back, we can conclude that that first episode of ALF was rather determined by the sorbitol contained in the acetaminophen syrup and secondary to bacterial infection with Pseudomonas aeruginosa.
There are two disorders of fructose metabolism in children, both of them with autosomal recessive transmission: HFI and 1,6-biphosphatase (FBP1) deficiency. FBP1-deficiency is considered an inborn error of fructose metabolism even though it is not a part of fructose metabolism pathway, being an important step in gluconeogenesis. HFI is caused by mutations in the ALDOB gene, involved in aldolase B synthesis. The disorder is due to aldolase B deficiency, an enzyme found in the liver, kidney or small intestine. Aldolase B plays a key role in carbohydrate metabolism (the glycolytic-gluconeogenic pathway), being involved in two reactions from the second path of fructose metabolism. Initially, it is responsible for cleaving the fructose-1-phosphate (F-1-P) molecule into D-glyceraldehyde and dihydroxyacetone phosphate and, to a lesser degree, in the breakdown of the fructose-1,6-biphosphate to D-glyceraldehyde-3-phosphate and dihydroxyacetone phosphate (Figure 2). These reactions are part of the processes of glycolysis and gluconeogenesis, their damage causing hypoglycemia and metabolic acidosis. On the other hand, the absence of the aldolase-B leads to the accumulation of F-1-P in the liver or renal tubular cells and to an increase of free fructose in the blood(3).
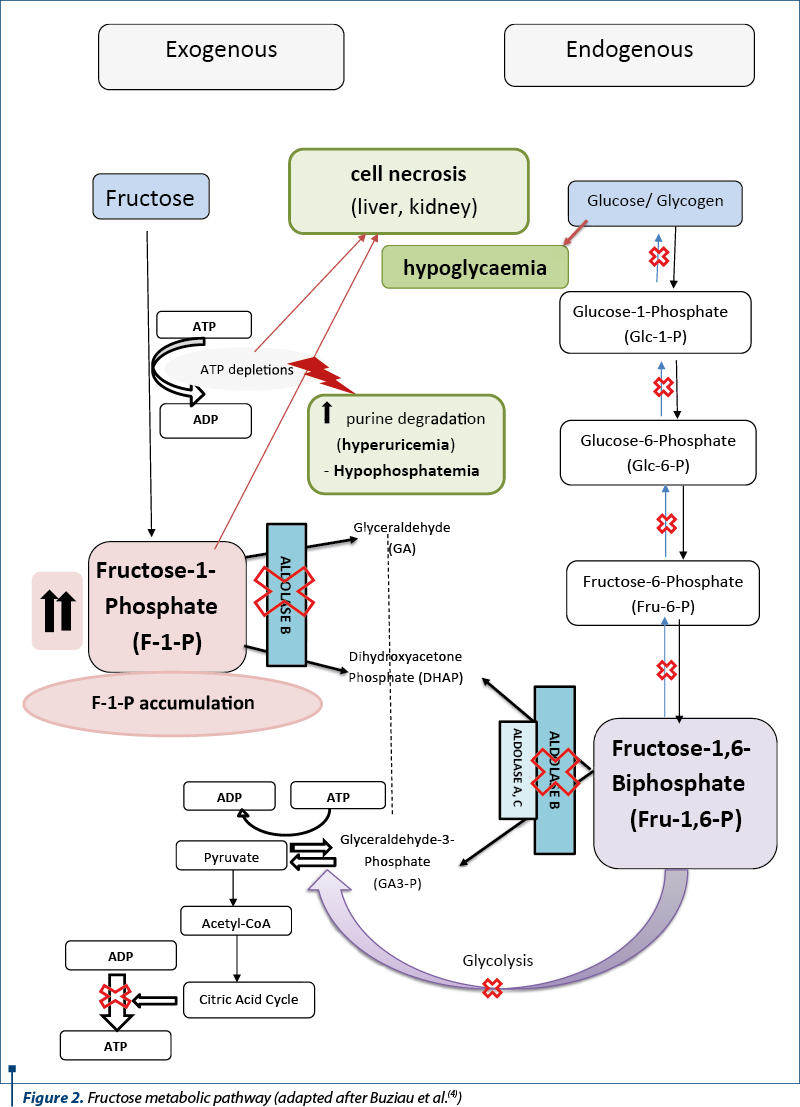
The clinical manifestations in HFI occur when fructose- and sucrose-containing foods are introduced into the diet of infants, often around the age of 4-6 months. The most important symptoms are vomiting, sweating, abdominal pain, jaundice, hepatosplenomegaly, hepatocellular insufficiency, bleedings, hypoglycemia, hypotonia and seizures. The liver is the main affected organ. The early histopathological aspect is represented by the fatty degeneration of the hepatocytes and bile duct proliferation(5). The laboratory findings include hypoglycemia, metabolic acidosis with an increased anion gap, coagulation disorders, anemia, thrombocytopenia, hyperuricemia, hypermagnesemia and hyperalaninaemia(1,6). Hyperuricemia is attributed to the breakdown and increased turnover of adenosine. The depletion of ATP leads to an increased breakdown of AMP, which causes increased level of uric acid. The accumulation of fructose-1-phosphate and phosphate trapped intracellulary is responsible for hypophosphatemia(6). In kidney damage (renal Fanconi syndrome), tubular acidosis, hypophosphatemia or aminoaciduria are detected(3). Urinary chromatography or urinary NMR spectroscopy (a method that we use in other suspicions of metabolic disorders) may detect fructosuria secondary to the ingestion of substances containing fructose(7,8). The aldolase-B activity can be determined only from liver or intestinal tissue, a method that is not exactly easy. At present, molecular genetic testing is used for a certain diagnosis because it is noninvasive and has a high sensitivity. There are more than 50 mutations in the ALDOB gene, the most common being A149P, A174D, or N334K. In our case, the patient is homozygote for A175D mutation in the ALDOB gene, a mutation found in 11% of patients with HFI from Europe(9).
A dietary restriction of fructose, sucrose and sorbitol is necessary for the treatment of children with HFI. Once the condition is detected, the strict adherence to a restrictive diet consisting of the substitution of fructose with other carbohydrate sources (glucose, maltose or cornstarch) will improve the clinical picture and normalize the laboratory parameters. On the other hand, the noncompliance with the dietary recommendations will lead to chronic liver and/or renal disease, cataract or growth retardation. During decompensation periods, these children require glucose (dextrose) infusions, metabolic acidosis correction and supportive treatment of ALF(1-3). The prognosis in acute decompensation depends on several factors, among which encephalopathy degree, INR or coagulation factors V or VII(10,11). Extremely important in HFI is the knowledge and avoidance of ingredients commonly used as sweeteners, such as honey, high-fructose corn syrup, agave syrup, inverted sugar, maple-flavored syrup, molasses, palm or coconut sugar or sorghum, which should be avoided. Also, a special attention is needed regarding some medications and especially syrups in which fructose, sucrose or sorbitol may represent primary components. Moreover, the sorbitol arginine solution, which is frequently used in the treatment of hepatic encephalopathy in patients with ALF, is contraindicated in HFI, due to the high sorbitol content. The long-term prognosis of these patients is good, with the elimination of fructose, sucrose and sorbitol from the diet, requiring periodic evaluations of growth rate, of liver and renal function, blood glucose or serum electrolytes(1-3,12).
Conclusions
We chose to present this case with HFI due to the early onset of the disease at a very young age (7 weeks), a long time before the introduction of fruit juices in the child’s diet. The sucrose from the most syrups for children can be the trigger for HFI, causing severe forms of the disease, sometimes life-threatening. The acute episode of HFI at this age may be confounded with sepsis, infectious hepatitis, or other IEM, such as galactosemia. The early diagnosis of HFI is important in order to begin the specific diet and to assure a good long-term prognosis.
Bibliografie
-
Baker PII, Ayres L, Gaughan S, et al. Hereditary Fructose Intolerance. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK333439
-
Hegde VS, Sharman T. Hereditary Fructose Intolerance. 2020 Oct 1. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2020; PMID: 32644528.
-
Sansaricq C, Balwani M. Disorders of fructose Metabolism. In: Sarafoglou K, Hoffmann GF, Roth KS (editors): Paediatric endocrinology and inborn of metabolism. USA. McGraw Hill Medical, 2008; 135-139.
-
Buziau AM, Schalkwijk CG, Stehouwer CD, et al. Recent advances in the pathogenesis of hereditary fructose intolerance: implications for its treatment and the understanding of fructose-induced non-alcoholic fatty liver disease. Cell Mol Life Sci. 2020;77:1709–1719.
-
Grama A, Burac L, Cainap SS, et al. Acute liver failure in children: aetiology and evolution. Arch Dis Child. 2019,104(S3):A1-A428.
-
Hegde VS, Sharman T. Hereditary Fructose Intolerance. Treasure Island (FL): StatPearls Publishing; 2020. Available at: https://www.ncbi.nlm.nih.gov/books/NBK559102
-
Grama A, Blaga L, Nicolescu A, et al. Novel Mutation in GALT Gene in Galactosemia Patient with Group B Streptococcus Meningitis and Acute Liver Failure. Medicina (Kaunas). 2019;55(4):91.
-
Grama A, Pop I, Tita G, et al. Galactosemia presented as a fulminant liver failure and Group b Streptococcus (GBS) sepsis. Archives of Disease in Childhood. 2017;102:A81.
-
Santer R, Rischewski J, Von Weihe M, Niederhaus M, et al. The spectrum of aldolase B (ALDOB) mutations and the prevalence of hereditary fructose intolerance in Central Europe. Hum Mutat. 2005;25(6):594.
-
Grama A, Aldea CO, Burac L, et al. Etiology and Outcome of Acute Liver Failure in Children – The Experience of a Single Tertiary Care Hospital from Romania. Children. 2020; 7: 282.
-
Grama A, Burac L, Aldea CO, et al. Vitamin D-Binding Protein (Gc-Globulin) in Acute Liver Failure in Children. Diagnostics. 2020;10:278.
-
Gonzalez-Anleo C, Gomez A, Gonzalez-Barcia M, et al. DI-089 New approach to the management of the hereditary fructose intolerance hypoglycaemia: Treatment with oral mannose. European Journal of Hospital Pharmacy. 2016;23:A157-A158.

Sindromul de regresie caudală – afecţiune congenitală rară cu implicaţii sistemice multiple
Ioana Oprescu, Cristina Becheanu, Ruxandra Darie, Mirela Pavelescu
Caudal regression syndrome (CRS) – also known as caudal dysplasia, sacral agenesis or sacral defect with anterior meningocele – is a rare congenital condition of unknown cause which consists in a developmental defect of ...
Studiu prospectiv privind managementul durerii acute la nou-născutul cu afecţiuni chirurgicale prin corelaţia scalei de durere CRIES cu protocolul terapeutic etapizat
Valentin Munteanu, Ioana Alecsandra Munteanu, Iulia Ciongradi, Ioan Sârbu, Elena Hanganu, Bogdan A. Stana, Maria Stamatin
The last 2-3 decades have brought major changes in the management of pain in the newborn....
Elemente actuale privind fiziopatologia cirozei hepatice la copil
Bogdan A. Stana, Evelina Moraru, Alice Azoicăi, Valentin Munteanu, Doina Mihăilă, Roxana Mihaela Barbu
Liver cirrhosis is a serious symptomatic complex, caused by progressive and generally irreversible liver damage, which causes the destruction of the lobular architecture by extensive fibrosis, nodular regeneration, and i...
Sindromul de regresie caudală – afecţiune congenitală rară cu implicaţii sistemice multiple
Ioana Oprescu, Cristina Becheanu, Ruxandra Darie, Mirela Pavelescu
Caudal regression syndrome (CRS) – also known as caudal dysplasia, sacral agenesis or sacral defect with anterior meningocele – is a rare congenital condition of unknown cause which consists in a developmental defect of ...
Particularități evolutive la doi pacienți cu atrezie de căi biliare – prezentare de cazuri
Gabriel Benţa, Alina Grama, Tudor Lucian Pop
Atrezia biliară (ACB) face parte din categoria colangiopatiilor mediate imunologic, alături de colangita sclerozantă neonatală şi de colangita sclerozantă primitivă, afecţiuni inflamatorii progresive şi severe, care...