Pulmonary hypertension (PH) defines an increase in mean pulmonary arterial pressure (PAPm) at a value equal to or greater than 25 mmHg, determined invasively by right cardiac catheterization. The hemodynamic definition of PH considers the combination of different hemodynamic parameters: mPAP, pulmonary wedge pressure (PWP), cardiac output (CO), diastolic pressure gradient (DPG) and pulmonary vascular resistance (PVR). The clinical correspondence of the haemodynamic definition, considering both the pathological aspect and the therapeutic approach, classifies the clinical conditions that associate PH in five clinical groups. Worldwide data on the incidence of pulmonary hypertension are not systematically reported; data on PH prevalence in different aetiologies are not published in detail. PH epidemiology documentation becomes necessary in order to standardize diagnostic and therapeutic management practices, with particular interest in aligning the differences between different regions of the world, not only geographically but also economically. The only data on the survival rate of PH patients in Eastern Europe are published from the Czech Republic registry. The better survival in the thromboembolic PH subgroup may be positively influenced by the availability of specific pharmacological therapy since 2007. The Portuguese registry reported in 2017 a survival rate at 1, 3 or 5 years of 95%, 77% and 71%, respectively, for new cases with different PH aetiologies, and the absence of statistically significant differences between different aetiology groups, but with a better prognosis for congenital heart disease related PH cases. For Romania, the National Pulmonary Hypertension Register has centralized the data from the six national diagnostic and treatment centres by 2017. The PH pathophysiology is based on pulmonary vascular remodelling by endothelial cell proliferation, smooth muscle cell hypertrophy and perivascular inflammation.
Hipertensiunea pulmonară – consideraţii generale
Pulmonary hypertension – general considerations
First published: 28 octombrie 2021
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Med.143.5.2021.5538
Abstract
Rezumat
Hipertensiunea pulmonară (HTP) defineşte o creştere a presiunii arteriale pulmonare medii de repaus (PAPm) la o valoare egală sau mai mare de 25 mmHg, determinată invaziv prin cateterism cardiac drept. Definiţia hemodinamică a HTP consideră evaluarea în asociere a diferiţi parametri hemodinamici: PAPm, presiunea pulmonară blocată (PWP), debitul cardiac (CO), gradientul de presiune diastolic (DPG) şi rezistenţa vasculară pulmonară (PVR). Corespondenţa clinică a definiţiei hemodinamice, considerând şi aspectul patologic, dar şi abordarea terapeutică, clasifică afecţiunile clinice care asociază HTP în cinci grupuri clinice. La nivel mondial, datele privind incidenţa hipertensiunii pulmonare nu sunt sistematic raportate, informaţii cu privire la prevalenţa HTP în diferite etiologii nefiind publicate unitar şi detaliat. Documentarea epidemiologiei HTP devine necesară în scopul uniformizării practicilor de diagnostic şi management terapeutic, cu interes deosebit în alinierea diferenţelor între regiunile lumii, diferite nu doar geografic, dar şi economic. Singurele date cu privire la rata de supravieţuire a pacienţilor cu HTAP din Europa de Est sunt publicate din registrul Republicii Cehe. Supravieţuirea mai bună a cazurile din subgrupul cu HTP tromboembolică poate fi influenţată pozitiv prin disponibilitatea terapiei farmacologice specifice, începând cu anul 2007. Registrul portughez de HTP raportează în 2017, pentru cazurile noi cu etiologii diferite de HTAP, o supravieţuire la 1, 3 sau 5 ani de 95%, 77%, respectiv 71% şi absenţa diferenţelor semnificative statistic între grupurile de etiologii diverse, dar cu un prognostic mai bun pentru cazurile de HTAP asociată bolilor cardiace congenitale. Pentru România, în Registrul Naţional de Hipertensiune Pulmonară au fost centralizate datele din cele şase centre naţionale de diagnostic şi tratament până în anul 2017. Fiziopatologia HTAP are la bază remodelarea vasculară pulmonară prin proliferare endotelială celulară, hipertrofia celulelor musculare netede şi inflamaţie perivasculară.
Hipertensiunea pulmonară (HTP) defineşte o creştere a presiunii arteriale pulmonare medii de repaus (PAPm) la o valoare egală sau mai mare de 25 mmHg, determinată invaziv prin cateterism cardiac drept(1). Definiţia hemodinamică a HTP consideră evaluarea în asociere a diferiţi parametri hemodinamici: PAPm, presiunea pulmonară blocată (PWP), debitul cardiac (CO), gradientul de presiune diastolic (DPG) şi rezistenţa vasculară pulmonară (PVR). Corespondenţa clinică a definiţiei hemodinamice, considerând şi aspectul patologic, dar şi abordarea terapeutică, clasifică afecţiunile clinice care asociază HTP în cinci grupuri clinice(1).
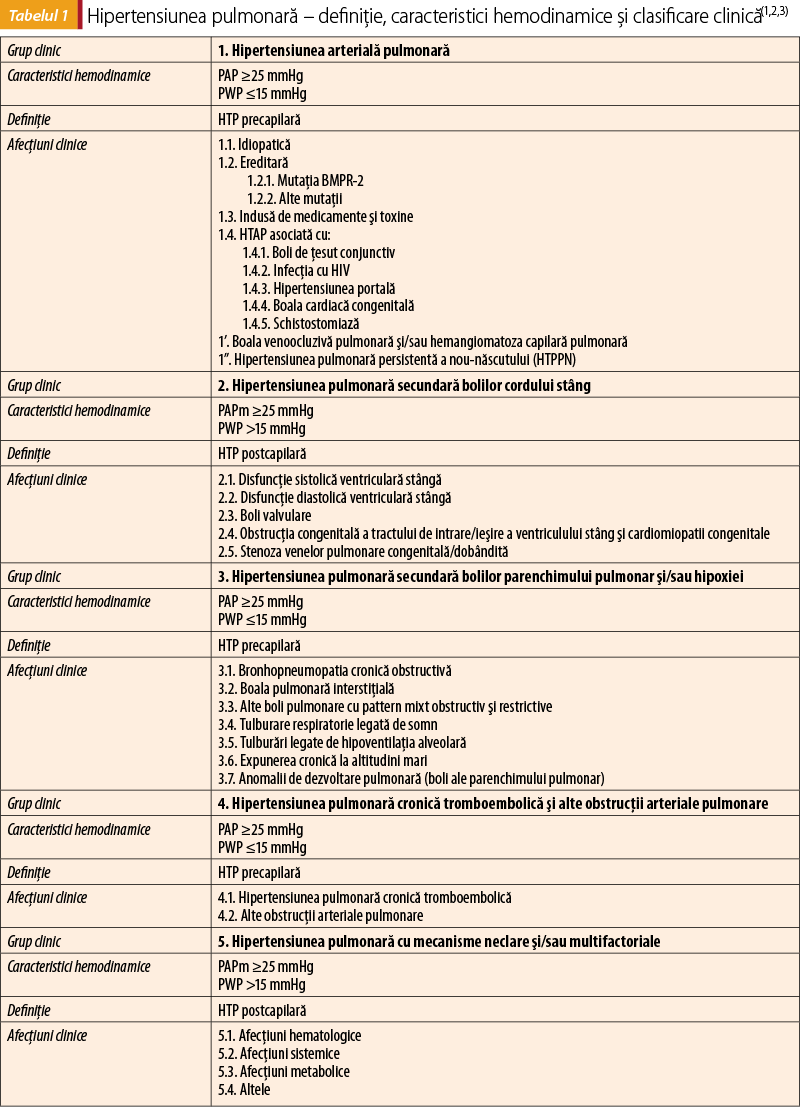
Grupul 1.2 al HTAP – hipertensiunea pulmonară ereditară – include mutaţii genetice ale genei BMPR-2 (mai frecvente) şi mutaţii genetice recente (detaliate în capitolul privind genetica HTP).
Hipertensiunea arterială pulmonară asociată bolilor cardiace congenitale cuprinde următoarele entităţi:
sindromul Eisemenger, caracterizat prin creşterea marcantă a PVR şi inversarea şuntului sistemico-pulmonar în şunt pulmono-sistemic sau şunt bidirecţional;
HTAP asociată cu şunturi sistemico-pulmonare, în care PVR are o creştere uşoară spre moderată şi include şunturile corectabile, dar şi cele necorectabile;
asociată cu defecte mici/incidentale, cu o creştere nemotivată şi severă a PVR asociată unor defecte mici în care corectarea defectului nu este indicată;
HTAP asociată cu bolile cardiace congenitale corectate chirurgical;
hipertensiunea pulmonară apărută sau persistentă în absenţa unor leziuni cardiace congenitale semnificative postoperatoriu(1).
Incidenţă şi prevalenţă
La nivel mondial, datele privind incidenţa hipertensiunii pulmonare nu sunt sistematic raportate, informaţii cu privire la prevalenţa HTP în diferite etiologii nefiind publicate unitar şi detaliat(1).
Date estimative cu privire la prevalenţa şi incidenţa HTAP în diferite ţări din Europa au fost publicate începând cu anul 1981 – primul registru de HTP primară din Marea Britanie. National Institutes of Health (NIH) a colectat datele din 32 de centre naţionale, totalizând 187 de pacienţi diagnosticaţi cu HTP primară. Datele demografice prezentau o vârstă medie de 36±15 ani, cu o prevalenţă a bolii raportată la sexul feminin versus masculin de 1,7:1. Datele de registru au urmărit manifestările clinice, remarcând o întârziere a diagnosticului şi prezentând o concluzie asupra importanţei diagnosticului precoce al hipertensiunii pulmonare primare(4).
La nivel global, documentarea epidemiologiei HTP devine necesară în scopul uniformizării practicilor de diagnostic şi management terapeutic, cu interes deosebit în alinierea diferenţelor între regiunile lumii diferite nu doar geografic, dar şi economic(5). Lansat în 2007, registrul COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension)(6), care cuprinde datele pacienţilor înrolaţi din Austria, Belgia, Germania, Italia, Olanda, Elveţia şi Maria Britanie, publică o analiză preliminară cu raportarea mediei de vârstă peste 65 de ani şi a caracteristicilor acestei grupe de vârstă în grupul de HATP idiopatică. Aparent, pentru grupul de HTP idiopatică, modelul demografic se modifică, afecţiunea fiind diagnosticată în măsură egală la ambele sexe, la vârste mai înaintate(7).
Registrul Francez de HTP a cuprins datele colectate din 17 spitale universitare, cu experienţa includerii pe durata unui an a unui număr minim de cinci pacienţi nou diagnosticaţi cu HTAP(8). Incidenţa HTAP a fost de 2,4 cazuri la 1 milion de locuitori adulţi/an, cu 75% dintre pacienţi în clasa funcţională NYHA III sau IV la momentul includerii în registru. Prevalenţa globală estimată în Franţa a fost de 15/1 milion de locuitori adulţi, cu o prevalenţă a HTAP idiopatice de 5,9 cazuri. The Scottish Pulmonary Vascular Unit (SPVU) a raportat în 2005 incidenţa anuală totală a HTAP înregistrată în Centrele Experte de 7,6 cazuri la 1 milion de locuitori; prevalenţa HTAP idiopatice a fost de 9 cazuri/1 milion locuitori, 10 cazuri/1 milion locuitori pentru HTP asociată cu boli de ţesut conjunctiv şi 7 cazuri de HTP asociată bolilor cardiace congenitale la 1 milion de locuitori(9).
Analiza datelor din 11 registre existente în şase ţări (SUA, Franţa, Spania, Scoţia, Anglia şi China) a fost publicată în 2013, prezentându-se concluzii importante legate de epidemiologia şi fenotipul pacienţilor cu HTAP. Sunt notabile schimbările semnificative în privinţa vârstei în creştere la momentul diagnosticului, a predominanţei diferite a sexului feminin în datele, dar şi creşterea ratei de supravieţuire(10).
Date mai recente cu privire la epidemiologia şi supravieţuirea pe termen lung din populaţia Europei de Est sunt publicate din Registrul Naţional al Republicii Cehe. Au fost documentate predominanţa hipertensiunii arteriale pulmonare la sexul feminin, vârsta medie de 51,9±16,9 ani şi incidenţa vârstei mai crescute comparativ cu vârsta cazurilor prevalente. Incidenţa anuală a HTAP a fost raportată ca fiind de 10,7 cazuri la 1 milion de locuitori cu o prevalenţă de 22,4 cazuri/1 milion de locuitori(11).
În 2015, Mueller-Mottet şi colaboratorii au publicat concluziile Registrului Elveţian de HTP în urma analizei datelor colectate în perioada 1998-2012. Începând cu anul 2000, incidenţa cazurilor cu vârstă mai avansată este mai crescută; cazurile incluse prezintă parametri hemodinamici mai buni în majoritatea cazurilor aflate sub tratament specific. Pacienţii diagnosticaţi cu HTAP şi hipertensiune arterială cronică tromboembolică (CTEPH) după anul 2008 au avut o supravieţuire mai bună comparativ cu cei diagnosticaţi anterior acestui an sau altei grupe de HTP(12).
De-a lungul anilor, modificările raportate în epidemiologia HTP pot fi influenţate de factori independenţi de boala în sine. Clasificarea nouă a HTP, extinderea examinării ecocardiografice Doppler, alături de conştientizarea şi utilizarea la scară mai largă a terapiilor specifice pot fi factori cauzatori ai acestor modificări. Conştientizarea de către profesionişti a existenţei acestei patologii la nivelul asistenţei medicale primare, dar nu numai, poate contribui în egală măsură la diagnosticul precoce, cu influenţă pozitivă asupra evoluţiei ulterioare a pacienţilor(13).
Pacienţii diagnosticaţi în perioada noiembrie 2015 – noiembrie 2016 cu HTAP sau CTEPH distală la Spitalul Universitar din Zürich (Elveţia) au fost evaluaţi comprehensiv, publicarea rezultatelor epidemiologice fiind făcută în aprilie 2018. 63% dintre cazurile investigate au fost diagnosticate cu HTAP idiopatică şi 20% cu HTAP asociată bolilor de colagen. Pentru întregul lot de pacienţi cu HTAP, mediana de vârstă a fost mai mică (55 de ani) comparativ cu mediana de vârstă (67 de ani) pentru cazurile diagnosticate cu CTEPH(13).
Pentru România, în Registrul Naţional de Hipertensiune Pulmonară au fost centralizate datele din cele şase centre naţionale de diagnostic şi tratament până în anul 2017.
Supravieţuire şi prognostic
Hipertensiunea pulmonară este o afecţiune cu o rată crescută a mortalităţii. Registrele care colectează date (definite conform The European Medicines Agency(14) pentru Europa sau potrivit The Agency for Healthcare Research and Quality pentru SUA(15)) au ca obiectiv frecvent urmărit supravieţuirea pacienţilor cu HTP(10). Datele publicate în 1991 pe un lot de 104 pacienţi diagnosticaţi cu HTAP idiopatică în SUA notează o mediană de supravieţuire de 2,8 ani, cu o rată de supravieţuire la cinci ani de 34%. Analiza a cuprins variabile clinice, hemodinamice şi variabile funcţionale pulmonare, fără evaluarea terapiei(16).
Singurele date cu privire la rata de supravieţuire a pacienţilor cu HTAP din Europa de Est sunt publicate 23 de ani mai târziu din Registrul Republicii Cehe. Comparativ, s-a demonstrat o supravieţuire la trei ani de 74% în populaţia incidentă diagnosticată cu HTAP. Pentru subgrupul de HTAP idiopatică/ereditară, supravieţuirea la 1, 2 sau 3 ani a fost mai scăzută, iar în subgrupul de HTAP asociată bolilor cardiace congenitale (1.4.4) a fost de 96% la trei ani. Supravieţuirea mai bună a cazurile din subgrupul 1.4.4 poate fi influenţată pozitiv prin disponibilitatea terapiei farmacologice specifice, începând cu anul 2007, fapt ce a condus la o referire mai bună a cazurilor către centrele experte(11).
Registrul REVEAL (Registry to Evaluate Early and Long-term PAH Disease Management) a cuprins datele observaţionale din 55 de centre din SUA şi a demonstrat o rată scăzută de supravieţuire la cinci ani, în ciuda progreselor marcante în terapia specifică a HTAP. Rata de supravieţuire la un an a pacienţilor cu diagnostic de HTAP anterior includerii în Registrul REVEAL a fost mai bună comparativ cu rata de supravieţuire a pacienţilor nou diagnosticaţi cu HTAP şi incluşi în urmărire. Pentru aceleaşi caracteristici analizate, supravieţuirea la cinci ani se menţine cu diferenţe procentuale de supravieţuire identice în favoarea cazurilor cu antecedente cunoscute de HTAP (supravieţuire de 65,4% comparativ cu 61,2% rată de supravieţuire a pacienţilor fără anamneză diagnostică)(17).
Datele obţinute din cea mai mare cohortă de pacienţi cu HTP înrolaţi într-un singur centru expert, Spitalul Universitar Giessen (Germania), au relevat o rată de supravieţuire semnificativ diferită între diferitele etiologii ale HTP. Pentru pacienţii din grupul 1 – HTAP, supravieţuirea la 1, 3 sau 5 ani a fost semnificativ mai bună pe fiecare durată de timp analizată, comparativ cu supravieţuirea pacienţilor din grupul 3 – HTP secundară bolilor parenchimului pulmonar şi/sau hipoxiei(18).
Registrul Portughez de HTP raportează în 2017, pentru cazurile noi cu etiologii diferite de HTAP, supravieţuire la 1, 3 sau 5 ani de 95%, 77%, respectiv 71% şi absenţa diferenţelor semnificative statistic între grupurile de etiologii diverse, dar cu un prognostic mai bun pentru cazurile de HTAP asociată bolilor cardiace congenitale(19).
Estimată ca o afecţiune cu o prevalenţă scăzută, datele epidemiologice pentru grupul 4 de HTP (CTEPH) sunt relativ limitate. În cazul diagnosticului de CTEPH într-un interval de doi ani de la evenimentul tromboembolic pulmonar, a fost raportată o prevalenţă de 3,8-4,8% din cazuri(20). Incidenţa CTEPH după un episod tromboembolic acut a fost de 0,57%, detectată într-un program de screening(21). Mortalitatea cazurilor de CTEPH a prezentat o descreştere semnificativă odată cu extinderea procedurilor chirurgicale de trombendarterectomie. Mortalitatea la un an, raportată în Registrul European de CTEPH, a fost sub 20%.
Pentru HTAP ereditară cu mutaţie a genei BMPR2, supravieţuirea este scăzută şi modificările hemodinamice sunt mai severe la pacienţii tineri, având un risc mai crescut de deces sau de transplant pulmonar comparativ cu cazurile fără mutaţie genetică(22).
Fiziopatologia hipertensiunii pulmonare. Evaluare şi prognostic
Mecanisme moleculare
Hipertensiunea pulmonară reprezintă manifestarea comună finală a numeroase afecţiuni şi sindroame. O serie de mediatori moleculari şi căi de semnalizare moleculară sunt implicate în fiziopatologia HTP. Modificările fiziopatologice în HTP sunt complexe pentru fiecare grup clinic, asociate cu un punct comun ce cuprinde o proliferare excesivă a celulelor musculare netede în arterele pulmonare (PASMC) în stratul medial, având ca rezultat final o supraîncărcare de postsarcină a ventriculului drept(23,24). Disfuncţia ventriculului drept datorată miocardului ischemic sau hibernant, ca o cauză majoră de deces sau de alterare a stării clinice a pacienţilor cu HTAP, poate fi considerată o ţintă terapeutică(25).
Disfuncţia endotelială şi remodelarea arterială consecutivă proliferării celulare a musculaturii netede sunt mecanisme similare implicate în etiologia proceselor tumorale, procese accelerate de proliferare în analogie cu apoptoză limitată, un metabolism glicolitic la nivelul musculaturii netede arteriale, fibroblaştilor şi al celulelor endoteliale(26).
Scăderea bioactivităţii oxidului nitric (NO) şi a prostaciclinelor, concomitent cu stimularea endotelinei şi a tromboxanilor, creează un dezechilibru ce generează disfuncţie endotelială prin favorizarea vasoconstricţiei, trombozei şi mitogenezei. Intens studiate în fiziopatologia HTAP, prostaciclinele prin intermediul AMPc acţionează ca un puternic vasodilatator pulmonar, inhibând activitatea plachetară şi proliferarea celulelor musculare netede (prin creşterea activităţii AMPc)(27).
Studiile asupra polimorfismului sintetazei endoteliale de oxid nitric din leziunile plexiforme de la nivelul patului vascular caracteristice HTAP, asociat cu boli cardiace congenitale, au identificat o creştere locală a NO(28). În HTP idiopatică sunt suspectate două cauze generatoare de reducere a NO: un metabolism intens al NO(29), alternativ cu o reducere a activităţii sintetazei endoteliale de NO, prin creşterea endogenă a inhibitorului sintetazei endoteliale NO, respectiv metabolismul proteic al dimetil-argininei asimetrice (ADMA). Datele au fost recent confirmate prin demonstrarea concentraţiilor scăzute de ADMA şi homocisteină ca mecanisme de adaptare la animale expuse unor condiţii de hipoxie. Inabilitatea homocisteinei şi a ADMA de activare a arginazei de tip II în condiţii de hipoxie conduce la o biodisponibilitate crescută a L-argininei pentru menţinerea sintezei de NO(30,31). Concentraţii plasmatice crescute de ADMA au fost identificate şi în HTP tromboembolică(32).
Sistemul endotelină (ET-1) este un alt puternic vasoconstrictor cu efect mitogenetic în proliferarea PASMC. Producţia crescută de ET-1 şi de tromboxan activează mecanisme de semnalizare care activează proliferarea PASMC. Descifrarea mecanismelor de vasoconstricţie exprimată şi a implicării mecanismului de disfuncţie endotelială va aduce o contribuţie importantă în strategiile de tratament al HTP(23).
În HTAP secundară bolilor cardiace congenitale, răspunsul imunologic alterat, răspunsul adrenergic crescut şi anormalităţi moleculare şi metabolice determină o transformare din statusul adaptativ al VD (hipertrofie VD compensată – HVDc), spre disfuncţie sistolică şi status maladaptiv al VD (hipertrofie de VD decompensată – HVDd)(33). Modificările din statusul HVDc pentru a trece în HVDd presupun stres oxidativ în exces, disfuncţie mitocondrială, insuficienţa aportului energetic şi transformarea metabolică spre glicoliză. Mecanismele de adaptare ale VD la supraîncărcarea prin presiune sunt determinante pentru supravieţuirea pacienţilor diagnosticaţi cu hipertensiune pulmonară secundară bolilor cardiace congenitale(34). Remodelarea VD, corelată cu anumite modificări metabolice moleculare, poate fi evaluată imagistic prin tomografia cu emisie de pozitroni (PET)(35).
Genetica hipertensiunii pulmonare
Dintre cazurile de HTAP ereditară, 75% prezintă mutaţii heterozigote ale genei BMPR-2, cuprinsă în marea familie a TGF-b, cu rol în controlul creşterii şi al tonusului vascular(1). Cu o prezenţă de 20-30% a acestei mutaţii şi în cazurile iniţial considerate ca HTPA idiopatică, a fost demonstrată existenţa unei corelaţii directe între deficitul de BMPR-2, răspunsul inflamator exagerat şi evoluţia spre HTAP(36).
Mutaţiile genei ACVRL1 (tip I receptor din familia TGF-b) şi ale genei ENG (tip II receptor accesor din familia TGF-b) sunt asociate cu hipertensiunea arterială ereditară întâlnită la pacienţii cu teleangiectazie hemoragică ereditară. În HTAP ereditară au fost identificate prin secvenţierea întregului exom la familii fără mutaţie cunoscută a BMPR-2 mutaţii ale genelor SMAD şi CAV1(37). Mutaţia heterozigotă a genei KCNK3, gena care codifică canalele de potasiu, determină apariţia HTAP ereditare prin mecanisme de alterare a funcţiei acestor canale(38).
Investigarea mutaţiilor genei EIF2AK4 a demonstrat un potenţial beneficiu în testarea genetică a acestei mutaţii pentru cazurile diagnosticate cu forme severe de HTP, în scopul diagnosticului diferenţial între HTAP şi hemangiomatoza capilară pulmonară familială sau boala venoocluzivă pulmonară familială(39).
Epigenetica HTP a identificat, dintre factorii de creştere şi de proliferare celulară a musculaturii netede vasculare pulmonare, factorul de creştere derivat din trombocite (PDGF), care joacă un rol esenţial în modificările vasculare cronice evidenţiate în HTP, alături de un potent rol mitogen exprimat în HTAP. Factorul de creştere endotelial vascular (VEGDF), o subfamilie a PDGF, a demonstrat, de asemenea, o creştere a expresiei în leziunile plexiforme ale HTAP şi o posibilă implicare în remodelarea vasculară indusă de hipoxie(23).
Parametrii clinici, imagistici
şi hemodinamici
Simptomatologia HTP nu poate fi definită ca o simptomatologie specifică acestui sindrom complex, fiind particulară fiecărei etiologii. Iniţial poate să includă epuizare fizică, dispnee, fatigabilitate, durere precordială anginoasă sau episoade sincopale. Progresia acestor fenomene asociate disfuncţiei de VD este marcantă în cazurile avansate. Semnele fizice ale HTP pot indica cauza imediată a HTP(1). Clasa funcţională OMS este un predictor puternic, utilizat în toate centrele experte în momentul diagnosticului, dar şi în evoluţia HTP(1).
O serie de variabile sunt constante în HTAP, nefiind modificate de terapia specifică: sex, vârsta, etiologia HTAP şi comorbidităţile individuale, fiecare dintre acestea având individual un potenţial predictiv scăzut (<5%), intermediar (5-10%) sau înalt (>10%). Deşi tratamentul specific HTAP se adresează factorilor de risc modificabili (simptomatologie, capacitate de efort fizic şi funcţia VD), impactul factorilor de risc nemodificabili este recomandat a fi cuantificat în stabilirea abordării terapeutice(40). Un studiu prospectiv aflat în desfăşurare urmăreşte identificarea variabilelor neinvazive şi invazive, care, asociate, reprezintă un pattern fidel de corelaţie cu supravieţuirea, dar şi cu obiectivele terapeutice în HTAP(41).
Evaluarea imagistică în HTP, de la stabilirea diagnosticului, a etiologiei, stratificarea riscului şi estimarea răspunsului la terapie, este esenţială(42).
Ecocardiografia cu urmărirea parametrilor VD pune accent pe importanţa evaluării complexe a unor parametri specifici hipertensiunii pulmonare. Siddiqui şi colab. au demonstrat recent importanţa evaluării unor parametri de strain longitudinal ai VD sau ai indicelui de excentricitate al VS, asociaţi semnificativ cu spitalizarea sau decesul(43).
Determinarea în dinamică a parametrilor hemodinamici ai VD, după iniţierea terapiei specifice la pacienţii cu HTAP, este importantă în evaluarea răspunsului terapeutic al VD. Cercetări recente orientează evaluarea parametrilor hemodinamici de performanţă ai VD către evaluarea indexului volum/bătaie (SVI), demonstrat ca variabilă independentă asociată cu prognosticul bolii, chiar şi atunci când debitul cardiac este normal(44). Registrul Giessen, în rezultatele publicate în 2017, indică faptul că testul de mers de 6 minute (6MWD) depăşeşte valoarea prognostică de supravieţuire, comparativ cu evaluarea de referinţă a clasei funcţionale la toate grupurile etiologice de HTP(18).
Evaluarea raportului ventilaţie/perfuzie (V/Q) a demonstrat sensibilitate şi specificitate comparabile cu cele ale explorării prin angio-computer tomografie a arterelor pulmonare în detectarea CTEPH(45).
Performanţa fizică de efort
Evaluarea obiectivă a performanţei fizice de efort utilizată la scară extinsă în centrele experte este realizată prin testul de mers de 6 minute (6MWD). Recomandările de ghid(1), precum şi rezultatele din studiile publicate(46) stabilesc un prag absolut de 380-440 de metri distanţă de mers, ca fiind asociat cu prognostic bun de supravieţuire. Publicate în 2010, rezultatele unei metaanalize a 22 de studii indică o lipsă de corelare între evenimentele clinice şi rezultatele 6MWD(47). Valoarea prognostică a acestei variabile utilizate în evaluarea pacienţilor cu HTP a demonstrat ulterior asocierea cu un prognostic negativ pentru distanţe de mers scăzute ale testului(44). Consecutiv, Zelniker şi colaboratorii au analizat în 2018 datele Registrului COMPERA(48) şi au demonstrat importanţa 6MWD în clinică şi pentru abordarea terapeutică a pacienţilor cu HTAP. Autorii subliniază importanţa informaţiei prognostice a creşterii distanţei de mers în analiza care a vizat prognosticul de mortalitate la un an comparativ cu analiza statistică a supravieţuirii pentru pacienţii diagnosticaţi cu HTAP.
Colective de cercetare au demonstrat şi susţin importanţa variabilelor obţinute prin testul de efort cardiopulmonar (CPET) în evaluarea progresiei HTAP şi în atingerea obiectivelor terapeutice(49,50). Consumul maxim de oxigen (peak VO2) şi pulsul de oxigen (VO2/frecvenţa cardiacă) au fost identificaţi ca predictori independenţi asociaţi cu supravieţuirea(51,52).
Medicaţie specifică
Strategiile terapiei specifice hipertensiunii pulmonare se adresează restabilirii balanţei dintre vasoconstricţie şi vasodilataţie, inversării remodelării anormale a patului vascular arterial, cu refacerea endoteliului vascular pulmonar. Concomitent cu utilizarea terapiei specifice, rata de supravieţuire a pacienţilor a crescut, aşa cum a fost prezentat anterior. A doua etapă de terapie, terapia specifică, se adresează terapiei iniţiale specifice, iar etapa a treia vizează abordarea terapeutică în concordanţă cu răspunsul la strategia iniţială(1). Indicaţiile de monoterapie şi obiectivele terapeutice curente în HTP sunt prezentate în figura 1.
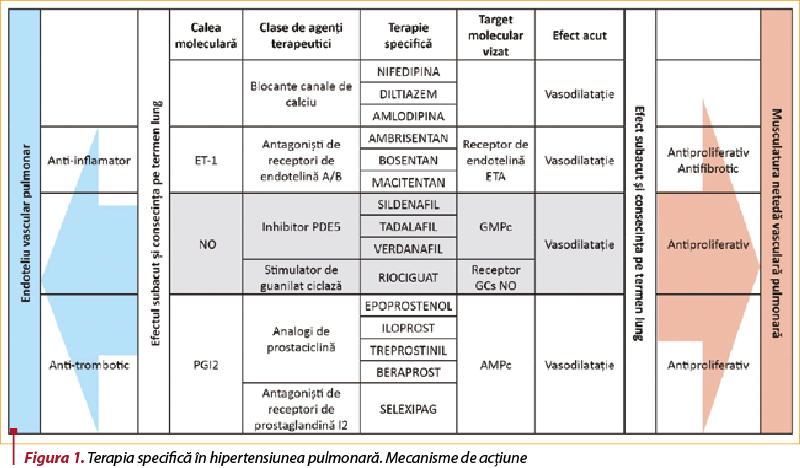
Terapia combinată, utilizată secvenţial sau direct, cu utilizarea simultană a două sau mai multe clase terapeutice, reprezintă o opţiune terapeutică viabilă. Introducerea medicaţiei specifice pentru HTAP (mono-, biterapie) a permis reducerea numărului de pacienţi cu indicaţie de transplant pulmonar sau a întârziat această procedură. Dar evoluţia pe termen lung a pacienţilor cu HTAP din clasele funcţionale III-IV OMS aflaţi sub tratament medical specific, inclusiv prostanoizi parenterali, nu este bine cunoscută şi, de aceea, transplantul pulmonar reprezintă o opţiune terapeutică viabilă (care trebuie luată în considerare). Metoda ECMO reprezintă o variantă de tratament temporar până la momentul transplantului pulmonar la pacienţii cu HTAP severă(53).
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67-119.
-
Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):5S-12S.
-
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) - web addenda The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and of the European Respiratory Society (ERS). Eur Heart J. 2016;37(1):67-119.
-
Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216-23.
-
Mocumbi AO, Thienemann F, Sliwa K. A global perspective on the epidemiology of pulmonary hypertension. Can J Cardiol. 2015;31(4):375-381.
-
Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation. 2014;129(1):57-65.
-
Pittrow D, Ghofrani HA, Opitz CF, Huscher D, Hoeper MM. International, prospective register for the documentation of first-line and maintenance therapy in patients with pulmonary hypertension (CompERA-XL). Dtsch Med Wochenschr. 2009;134 Suppl 5:S173-175.
-
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France. Results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023-1030.
-
Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30(1):104-109.
-
McGoon MD, Benza RL, Escribano-Subias P, Jiang X, Miller DP, Peacock AJ, et al. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol. 2013;62(25 Suppl):D51-59.
-
Jansa P, Jarkovsky J, Al-Hiti H, Popelova J, Ambroz D, Zatocil T, et al. Epidemiology and long-term survival of pulmonary arterial hypertension in the Czech Republic: a retrospective analysis of a nationwide registry. BMC Pulm Med. 2014;14:45.
-
Mueller-Mottet S, Stricker H, Domenighetti G, Azzola A, Geiser T, Schwerzmann M, et al. Long-term data from the Swiss pulmonary hypertension registry. Respiration. 2015;89(2):127-140.
-
Bartenstein P, Saxer S, Appenzeller P, Lichtblau M, Schwarz EI, Ulrich S. Risk factor profiles achieved with medical therapy in prevalent patients with pulmonary arterial and distal chronic thromboembolic pulmonary hypertension. Respiration. 2018;96:127–137.
-
European Medicines Agency. Guidelines on Pharmacovigilance for Medicinal Products for Human Use. https://ec.europa.eu/health/documents/eudralex/vol-9_en.
-
Gliklich RE, Dreyer NA, Leavy MB (ed). Registries for Evaluating Patient Outcomes: A User's Guide [Internet]. 3rd edition. Rockville (MD): Agency for Healthcare Research and Quality (US); 2014. Report No.:13(14)-EHC111. AHRQ Methods for Effective Health Care.
-
D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343-349.
-
Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, et al. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148(4):1043-1054.
-
Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, et al. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36(9):957-967.
-
Marques-Alves P, Baptista R, Marinho da Silva A, Pêgo M, Castro G. Real-world, long-term survival of incident patients with pulmonary arterial hypertension. Rev Port Pneumol. 2017;23(3):124-131.
-
Guérin L, Couturaud F, Parent F, Revel MP, Gillaizeau F, Planquette B, et al. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb Haemost. 2014;112(3):598-605.
-
Klok FA, van Kralingen KW, van Dijk AP, Heyning FH, Vliegen HW, Huisman MV. Prospective cardiopulmonary screening program to detect chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. Haematologica. 2010;95(6):970-975.
-
Evans JD, Girerd B, Montani D, Wang XJ, Galiè N, Austin ED, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. 2016;4(2):129–137.
-
Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443–455.
-
Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S-24S.
-
Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121(18):2045-2066.
-
Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med. 2014;20:191-201.
-
Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26(2):194–201.
-
Berger RM, Geiger R, Hess J, Bogers AJ, Mooi WJ. Altered arterial expression patterns of inducible and endothelial nitric oxide synthase in pulmonary plexogenic arteriopathy caused by congenital heart disease. Am J Respir Crit Care Med. 2001;163(6):1493-1499.
-
Demoncheaux EA, Higenbottam TW, Kiely DG, Wong JM, Wharton S, Varcoe R, et al. Decreased whole body endogenous nitric oxide production in patients with primary pulmonary hypertension. J Vasc Res. 2005;42(2):133-136.
-
López V, Moraga FA, Llanos AJ, Ebensperger G, Taborda MI, Uribe E Plasmatic concentrations of ADMA and homocystein in Llama (Lama glama) and regulation of arginase type II: An animal resistent to the development of pulmonary hypertension induced by hypoxia. Front. Physiol. 2018;9:606.
-
Kao CC, Wedes SH, Hsu JW, Bohren KM, Comhair SA, Jahoor F, et al. Arginine metabolic endotypes in pulmonary arterial hypertension. Pulm Circ. 2015;5(1):124-134.
-
Zhang S, Yang T, Xu X, Wang M, Zhong L, Yang Y, et al. Oxidative stress and nitric oxide signaling related biomarkers in patients with pulmonary hypertension: a case control study. BMC Pulm Med. 2015;15:50.
-
Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl). 2013;91(11):1315-1327.
-
Guimaron S, Guihaire J, Amsallem M, Haddad F, Fadel E, Mercier O. Current knowledge and recent advances of right ventricular molecular biology and metabolism from congenital heart disease to chronic pulmonary hypertension. Biomed Res Int. 2018;2018:1981568.
-
Amsallem M, Kuznetsova T, Hanneman K, Denault A, Haddad F. Right heart imaging in patients with heart failure: a tale of two ventricles. Curr Opin Cardiol. 2016;31(5):469-482.
-
Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(7):859-872.
-
Dodson MW, Brown LM, Elliott CG. Pulmonary arterial hypertension. Heart Fail Clin. 2018;14(3):255-269.
-
Bohnen MS, Roman-Campos D, Terrenoire C, Jnani J, Sampson KJ, Chung WK, et al. The impact of heterozygous KCNK3 mutations associated with pulmonary arterial hypertension on channel function and pharmacological recovery. J Am Heart Assoc. 2017;6(9). pii:e006465.
-
Best DH, Sumner KL, Smith BP, Damjanovich-Colmenares K, Nakayama I, Brown LM, et al. EIF2AK4 mutations in patients diagnosed with pulmonary arterial hypertension. Chest. 2017;151(4):821–828.
-
Weatherald J, Sitbon O, Humbert M. Validation of a risk assessment instrument for pulmonary arterial hypertension. Eur Heart J. 2017;0:1-4.
-
Prospective longitudinal study of patients with idiopathic pulmonary arterial hypertension, family or taking anorectics (EFORT). Disponibil la https://clinicaltrials.gov/ct2/show/NCT01185730.
-
Goerne H, Batra K, Rajiah P. Imaging of pulmonary hypertension: an update. Cardiovasc Diagn Ther. 2018;8(3):279-296.
-
Siddiqui I, Rajagopal S, Brucker A, Chiswell K, Christopher B, Alenezi F, et al. Clinical and echocardiographic predictors of outcomes in patients with pulmonary hypertension. Am J Cardiol. 2018;pii:S0002-9149(18)31191-3.
-
Weatherald J, Boucly A, Chemla D, Savale L, Peng M, Jevnikar M, et al. Prognostic value of follow-up hemodynamic variables after initial management in pulmonary arterial hypertension. Circulation. 2018;137(7):693–704.
-
Ley S, Ley-Zaporozhan J, Pitton MB, Schneider J, Wirth GM, Mayer E, et al. Diagnostic performance of state-of-the-art imaging techniques for morphological assessment of vascular abnormalities in patients with chronic thromboembolic pulmonary hypertension (CTEPH). Eur Radiol. 2012;22(3):607-616.
-
Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–172.
-
Savarese G, Paolillo S, Costanzo P, D'Amore C, Cecere M, Losco T, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol. 2012;60(13):1192-1201.
-
Zelniker TA, Huscher D, Vonk-Noordegraaf A, Ewert R, Lange TJ, Klose H, et al. The 6MWT as a prognostic tool in pulmonary arterial hypertension: results from the COMPERA registry. Clin Res Cardiol. 2018;107(6):460-470.
-
Taran I, Valieva Z, Martynyuk T, Chazova I. The value of cardiopulmonary exercise test in assessment of the severity of pulmonary arterial hypertension patients. Eur Respir J. 2017;50:PA2396.
-
Weatherald J, Farina S, Bruno N, Laveneziana P. Cardiopulmonary exercise testing in pulmonary hypertension. Ann Am Thorac Soc. 2017;14(Supplement 1):S84-S92.
-
Badagliacca R, Papa S, Valli G, Pezzuto B, Poscia R, Manzi G, et al. Echocardiography combined with cardiopulmonary exercise testing for the prediction of outcome in idiopathic pulmonary arterial hypertension. Chest. 2016;150(6):1313-1322.
-
Wensel R, Francis DP, Meyer FJ, Opitz CF, Bruch L, Halank M, et al. Incremental prognostic value of cardiopulmonary exercise testing and resting haemodynamics in pulmonary arterial hypertension. Int J Cardiol. 2013;167(4):1193–1198.
-
Bartolome SD, Torres F. Severe pulmonary arterial hypertension: stratification of medical therapies, mechanical support, and lung transplantation. Heart Fail Rev. 2016;21(3):347-356.
Articole din ediţiile anterioare
Managementul pacienţilor cu boală diareică în cabinetul medicului de familie
Diareea este una dintre cele mai frecvente boli ale sistemului gastrointestinal, având un efect major asupra calităţii vieţii. Sistemul digestiv r...
Depresia cu risc suicidar
Din cauza creșterii ratei de suicid prin depresie, această tulburare psihică a devenit una din cele mai studiate condiții psihiatrice.
Hipertensiunea pulmonară arterială - estimarea riscului și evaluare prognostică
Hipertensiunea pulmonară (HTP) este o condiţie fiziopatologică care include numeroase situații clinice şi care prin evoluția însăși a patologiei ...
De la ghiduri la practica clinică: recomandări actuale pentru abordarea pacientului cardiac cu patologie chirurgicală noncardiacă
Intervenţiile chirurgicale noncardiace (ICNC) reprezintă aproape 85% din totalul de 300 de milioane de intervenţii chirurgicale care sunt efectuat...