Castration-resistant prostate cancer(CRPC) is an important aspect of our every day clinical practice. For a very long period of time, the only treatment option available for fit patients was chemotherapy with Docetaxel associated with Prednisone. The recent approval in our country of new therapies like Abiraterone, will offer new treatment options which will enhance disease control and safety profile for selected patients. We have reviewed over 50 articles published in international journals, to offer an extensive view of therapeutic agents used in the management of CRPC, from chemotherapy agents, hormonal treatment, to new androgen receptor inhibitors, immunotherapy and prevention and palliative treatment of bone metastasis and SRE(skeletal related events) with bisphosphonates, radiotherapy and radiopharmaceuticals.
Managementul cancerului de prostată rezistent la castrare
Management of castration-resistant prostate cancer
First published: 24 octombrie 2015
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/OnHe.32.3.2015.4315
Abstract
Rezumat
Cancerul de prostată rezistent la castrare (CRPC) este un aspect important al practicii noastre clinice în fiecare zi. Pentru o perioadă foarte lungă de timp, singura optiune de tratament disponibilă pentru pacienţi a fost chimioterapia cu docetaxel asociat cu prednison. Recenta aprobare în ţara noastră de noi terapii, precum Abiraterone, va oferi noi opţiuni de tratament, care va spori controlul bolii şi profilul de siguranţă pentru pacienţii selectaţi. Am revizuit peste 50 de articole publicate în reviste internaţionale, pentru a oferi o vedere largă asupra agenţilor terapeutici utilizaţi în gestionarea CRPC, de la agenţi chimioterapici, tratament hormonal, la noi inhibitori ai receptorilor androgeni, imunoterapie şi prevenţie şi tratamentul paliativ al metastazelor osoase şi SRE (manifestări osoase asociate) cu bifosfonaţi, radioterapie şi radiofarmaceutice.
Prostate cancer (PC) is one of the most common cancers in countries with high life expectancy because the risk gradually increases with age. The incidence of PC in the European Union is 78,9/100.000 inhabitants/year, and the mortality rate is 26/100.000 inhabitants/year. The average age at diagnosis is 71 years. In cases of death by non-cancer causes, autopsy revealed a localized PC in 8% of the men aged between 20 and 29 years and in 83% of the ones aged between 70-79 years.
Screening healthy men by means of prostate specific antigen (PSA) increases PC incidence and determines overdiagnosis. Subclinical forms of prostate cancer are common in men over 50 years old. According to recent studies, the effect of intense screening and early treatment on mortality rates remains controversial.
The best-known risk factors for PC are: age, family history (risk increases two times if a first degree relative suffers from PC), race (in the US, the incidence is higher in black people than in Caucasians), geographical location (low incidence in Asia, high incidence in Scandinavia and US) and nutrition (increased animal fat intake is a possible risk factor, but its role has not been fully established in the ethiology of PC).
In patients where PC is suspected, serum PSA should be assessed and a digital rectal examination (DRE) should be performed. Deciding whether to perform a prostate biopsy or not should be based on PSA level, DRE results, prostate size, race, age, comorbidities, patient’s will, and, if available, results from previous biopsies. The prostate biopsy should be guided by trans rectal echography and at least eight tissue samples should be collected.
In terms of histology, the vast majority of prostate neoplasms are adenocarcinomas (95%). 70% of the prostate adenocarcinomas arise in the peripheral zone, 20% in the transitional zone and 10% in the central zone.
A well-performed prostate biopsy allows the pathologist to assess the Gleason score, which is essential for determining prognosis and management.
In 1941, Huggins and Hudges discovered that prostate tumours are almost entirely hormone-dependent. Initially, hormone therapy (HT) was used in advanced stages of PC, when the patient had symptoms or metastases diagnosed by imaging studies.
Currently, androgen deprivation therapy (ADT) is the treatment of choice for all stages of PC; ADT is also used as a neoadjuvant or adjuvant treatment before or after radiotherapy.
ADT lowers both PSA values and tumour size, and long-term therapy leads to a clinically stable interval when PSA values are lower than 4 ng/ml. No hormonal therapy is superior to another!
Patients are monitored by means of serum PSA and regular imagistic studies. The duration of treatment response is, on average, between 12 and 18 months; 20% of the patients still have a complete response after 5 years.
However, a subpopulation of prostate tumour cells acquires resistance to anti-androgen therapies and becomes dominant, resulting in hormone resistance (resistance to castration).
Castration resistant prostate cancer
Definition
Castration resistant prostate cancer (CRPC) is defined as the stage where the disease progresses despite maintaining serum testosterone values at “castration” level (either by surgery or hormonal therapy), which is considered by most experts to be <50 ng/dL.
Diagnosis:
CRPC is diagnosed by one or more of the following criteria:
- increase in plasma PSA levels: most of the patients exhibit a rise in PSA even if the testosterone level is kept <50 ng/dL, which is considered the castration level. The transition to CRPC is defined as an increase in serum PSA of at least 0.2 ng/mL when compared to the previous assessment;
- increase in size of pre-existing lesions/metastases;
- the presence of new metastases.
The increase of PSA serum values, in spite of low plasma testosterone, represents the transition towards the hormone-resistant stage, which is considered lethal for most patients. Clinically, CRPC is diagnosed in the following cases: increasing PSA values in the absence of metastatic disease; newly-diagnosed bone metastases; presence of non-osseous metastases; soft tissue metastases with or without bone damage; node enlargement alone or with concurrent bone and/or visceral metastases. In some cases, new metastases can be diagnosed without increased PSA values. The disease can be symptomatic or asymptomatic. Extra-osseous metastatic disease can develop also in rare sites, such as adrenal gland, kidney, pancreas and brain.
Pathophysiology
In CP, androgens are produced by:
- Leydig testicular cells;
- cholesterol conversion through the cytochrome system;
- prostate tumour tissue (autocrine secretion of androgens).
The conversion of prostate cancer towards CRPC requires the development of adaptive pathways for transmitting the intracellular signal in an androgen-free environment.
The hormone-resistance mechanisms are divided into 6 categories:
- increased expression of enzymes involved in steroidogenesis - studies indicate that in CRPC, even extremely low (castrate) levels of androgens are enough for activating the androgen receptors, and thus for the survival of the tumour cell; moreover, bone metastases contain cells with intact enzymatic equipment, capable of converting adrenalin androgens into testosterone and dihydrotestosterone;
- increased expression of androgen receptors - which is considered a consequence of a genetic modification that promotes an increase in receptor sensibility for androgens, enhancing it at DNA level;
- androgen receptor mutations and changes in the specificity of the receptor for its ligand – this phenomenon results in receptor activation by non-steroid molecules and by anti-androgens (a possible explanation of the paradox effect seen in patients with CRPC when androgen deprivation therapy is discontinued);
- one of the most important mechanisms involved in the development of CRPC is the activation of other pathways for intracellular signal transmission, which can determine an increase in androgen receptor activity or co-activation of the receptor in the absence of androgens. This also includes the activation of other receptors, like the ones for the epidermal growth factor or the tyrosine-kinases receptors;
- alternative intracellular signalling pathways also represent a mechanism involved in the transition to CRPC. This activation can inhibit the apoptosis induced by anti-androgen therapy through Bc1-2 anti-apoptotic protein activation;
- studies have shown that a subpopulation of PC tumour cells is made of undifferentiated stem cells which do not express androgen receptors, and thus they do not depend on androgens for survival.
Treatment
The first step consists in clearly establishing if serum testosterone is actually at “castration” levels. Approximately 1% of the patients undergoing treatment with LH-RH analogues do not respond completely to the therapy and have increased testosterone levels
The second step of the CRPC therapy is deciding if the disease is metastatic or non-metastatic, a feature which will influence therapy.
A. Non-metastatic CRPC
In patients with non-metastatic CRPC, there is an increase in PSA levels, but comprehensive imaging evaluation cannot detect the presence of metastases.
For these patients (usually on androgen deprivation therapy, such as Gosorelin/Leuprolid Acetate), their current medication can be associated with an androgen receptor antagonist (such as Bicalutamide, Flutamide or Nilutamide), thus obtaining a double androgenic blockade. This regimen can delay disease progression or prevent the occurrence of metastases, though there is no definitive clinical evidence to support this outcome.
If the disease progresses following the LH-RH agonists/antagonists and androgen receptor antagonist combination, discontinuation of androgen receptor antagonists (ARA) can be considered. Following this measure, almost 20% of the patients will have a subsequent decrease of over 50% in plasma PSA levels. Nevertheless, usually, this response is short-lasting, 3-5 months, and it appears 4-6 weeks after therapy discontinuation.
After the beneficial effect of discontinuing ARA therapy has disappeared, when plasma PSA values are increasing once more, LH-RH agonists/antagonists therapy can be associated with:
- ARA - a different drug than the one previously used would be preferred;
- Ketoconazole - a non-specific inhibitor of secondary androgen production; its use requires hydrocortisone supplementation due to risk of adrenal insufficiency. This intervention is associated with a PSA decrease in 35-50% of the patients.
REMEMBER
As long as CRPC is considered non-metastatic, ADT is preferred - various drug combinations; initiating chemotherapy is not recommended.
B. Metastatic CRPC
Maintaining testosterone at castrate levels, even after PC transitions to CRPC, is considered gold standard both in the locally-advanced and the metastatic disease, because androgen receptor and the intracellular androgen levels remain significantly elevated even in hormone resistant PC patients.
Patients will continue the therapy with LH/RH agonists/antagonists. High levels of testosterone, over the “castration” limits, can have detrimental effects. Constant androgenic suppression increases survival for patients with CRPC.
If the LH/RH agonists/antagonists therapy was associated with ARA therapy before transition to CRPC occurs, discontinuing ARA treatment can have palliative effects such as decreasing pain intensity, increasing Hb values and decreasing serum PSA levels. Studies report that medication withdrawal was successful for the following agents: flutamide, bicalutamide, nilutamide, megestrol acetate, diethylstilbestrol, estramustine. The response to discontinuing the anti-androgen medication appears after 4-6 weeks, depending on the half-life of the drug. This response could be attributed to the conversion of the antagonist effect to an agonist one, and it would be connected to the anti-androgen’s half-life. For example, flutamide, with a half-life of 7-8 weeks, will determine a faster response than bicalutamide, which has a longer half-life. Almost 20% of the patients receiving a combined androgenic blockade will have a decrease in PSA values of over 50% if anti-androgen therapy is discontinued (the percentage of patients varying between 15% and 33%), although this response will persist for only 3-5 months.
If no response to ARA therapy interruption is recorded, another drug from the ARA class can be given in combination with the LH/RH agonist/antagonist. Historically, useful drugs included: diethylstilbestrol (DES) 1 + 3 mg/day, estramustine (the first agent indicated in CRPC) and other parenteral formulas which determined short-term response rates ranging from 24% to 42%.
10 mg/day of Prednisone has proved to have a palliative effect on symptoms for CRPC patients. Hydrocortisone 30-40 mg/day or Dexamethasone 0.5-40 mg/day treatment has a response rate of 16-59%. Most studies report that steroid therapy has demonstrated a decrease in PSA levels, which is associated with increased survival.
REMEMBER
Long-term oestrogen therapy is associated with an increased thromboembolic risk, even for transdermal drugs.
Androgen biosynthesis inhibitors
Ketoconazole is an non-specific inhibitor of secondary androgen synthesis, which includes both synthesis in the adrenal cortical and tumour androgen production, two mechanisms that are related to hormone resistance. It is an imidazole with antimitotic action (when in high doses), that inhibits the CYP17 and CYP11B1 complexes, which are responsible for the function of steroidogenesis enzymes and the modulation of retinoic acid metabolism. Ketoconazole 60-1200 mg/day, associated with hydrocortisone (to lower the risk of adrenal cortical failure), can determine a decrease in PSA values for 50-70% of the patients with response rates correlated with serum levels of androgens. Ketoconazole can also be associated with anti-androgen therapy, but its side effects make it difficult to be tolerated. When used alone, Ketoconazole must be associated with hydrocortisone to compensate the side-effects of blocking steroidogenesis and the occurrence of adrenal cortical failure.
Attention! Ketoconazole must be administered before meals or with juice, in order to ensure a highly acidic gastric environment, which is necessary for a complete absorption. Simultaneous therapy with proton pump inhibitors or H2 antagonists will reduce Ketoconazole absorption. Tumour progression during Ketoconazole therapy is, most often, secondary to increasing androstenedione and dihidroandrostendion (DHEA) sulphate, which favours alternative ways of hormonal metabolism.
Currently, Ketoconazole tends to be replaced with abiraterone acetate.
Abiraterone acetate (Zytiga®), a pregnenolone analogue, is a selective and irreversible inhibitor of steroidogenesis that inhibits cytochrome P450, thus reducing extragonadal testosterone synthesis (including testosterone precursors, dehydroepiandrosterone and androstenedione) in the adrenal gland and malignant cells from prostate cancer; it is more effective than Ketoconazole. The side effects are due to mineralocorticosteroids excess and include: hypertension, fluid retention, oedema, convulsions, headache, muscle weakness, diarrhoea, hypokalemia. In 2010, it was introduced in the treatment of CRPC after Docetaxel chemotherapy. Currently, Abiraterone acetate (1000 mg p.o./day) with prednisone (5 mg x 2/day) is approved as standard therapy in hormone-resistant prostate cancer. Also, Abiraterone has shown to relieve pain and improve the quality of life in patients who previously received chemotherapy.
Next-generation androgen receptor inhibitors
Enzalutamide (Xtandi®) shows a high affinity for blocking the androgen receptor (AR) and inhibits the activated receptor translocation and, implicitly, its binding to the DNA. Enzalutamide impairs AR co-activating factors recruitment and DNA transcription of AR. Enzalutamide therapy is not associated with adverse effects that are specific to the androgen receptors antagonists mentioned earlier.
The phase III study that led to approving enzalutamide for patients with progressive disease after Docetaxel has demonstrated an increase in survival with 4.8 months and a 37% decrease in mortality; also, enzalutamide was associated with an increase of progression-free survival from 2.9 months to 8.3 months. Recently, a phase III randomized study (PREVAIL) reported favourable results for enzalutamide (160mg/day) treatment in patients with CRPC and no prior chemotherapy. Enzalutamide therapy was associated with: 50% decrease of PSA levels (78% vs. 3%), decrease in soft tissues tumour size (59% vs. 5%), longer PFS and increased interval before chemotherapy initiation. Enzalutamide is approved as standard therapy in CRPC since 2012.
Currently, based on the example of enzalutamide, new molecules are tested for multiple effects on androgenic receptors: ARN-509 and galeteron (TOK-001), with multiple effects on AR, including its blocking and degradation.
Immunotherapy
Sipuleucel-T (Provenge®) is a type of cellular immunotherapy produced by combined transfection of an adenovirus (together with granulocytes and macrophage stimulating factor) in activated mononuclear cells collected from the patient through leukophoresis and stored ex vivo with a PAP fusion protein; these cells are then reinjected in the CRPC patient’s prostate. The therapy’s aim is to activate immune cells so that the body generates an anti-tumour, immune mediated, response. Sipuleucel-T has shown a survival advantage over placebo (25.8 vs. 21.7 months); nevertheless, in most patients, it does not modify DFS or PSA values. For these reasons, Sipuleucel-T therapy should, ideally, be followed by another treatment that allows the short-term control of the disease and enables long-term survival improvement. Sipuleucel-T is well tolerated; its side effects include: asthenia, flu syndrome, chills, fever, myalgia and headache, which resolve within 24 to 48 hours from the infusion.
Chemotherapy
Traditionally, prostate cancer is considered chemoresistant. Nevertheless, in 1996, Mitoxantrone was approved to palliate pain secondary to bone metastasis. Mitoxantrone is a synthetic hydroxyquinone, similar in structure with doxorubicin. Treatment with Mitoxantrone does not increase survival, but improves symptoms in metastatic CRPC patients. Standard regimen is 12 mg/m2 every 3 weeks; side effects include: vomiting (61%), asthenia (39%), alopecia (29%), anorexia (25%), neutropenia (20%) with febrile neutropenia in only 2% of the patients. Decrease in cardiac function was observed in 5-7% of the patients.
In 2004, Docetaxel is introduced in the therapy of CRPC. Docetaxel acts on the mitotic spindle, which binds to tubulin, stabilizes microtubule formation and inhibits de-polymerization. In a 70 mg/m2 dose every 3 weeks, in association with prednisone, it has become the current gold standard in CPRC treatment. This drug offers not only clinical benefits (symptoms palliation), but also delays disease progression and prolongs general survival from 16.5 months (with mitoxantrone/prednisone) to 18.2 months (using Docetaxel and prednisone). Docetaxel is recommended for CRPC patients with progressive disease associated with a high symptom burden. Docetaxel seems a reasonable option for patients with rapidly progressive disease that is documented by objective imaging changes. Associating Docetaxel to other cytostatics did not prove to be superior to mono-chemotherapy.
Attention! In 2014, it has been demonstrated that using Docetaxel therapy in the first line of treatment for PC patients with unfavourable prognostic factors, associated with anti-androgen therapy, gives better results when compared to anti-androgen therapy alone. Thus, associating Docetaxel with anti-androgen therapy increases median survival with 13.6 months in hormone-sensitive PC patients (phase III study E3805 at the National Cancer Institute). Survival benefits obtained by early Docetaxel chemotherapy (75 mg/m2) associated with anti-androgen therapy (57.6 months) compared to androgenic therapy alone (44 months) was considered unprecedented in prostate cancer. Moreover, the median time to clinical disease progression was significantly improved from 19.8 months, in the case of mono-therapy, to 32.7 months by Docetaxel association.
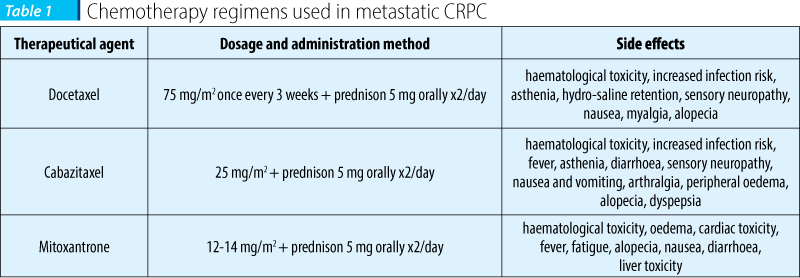
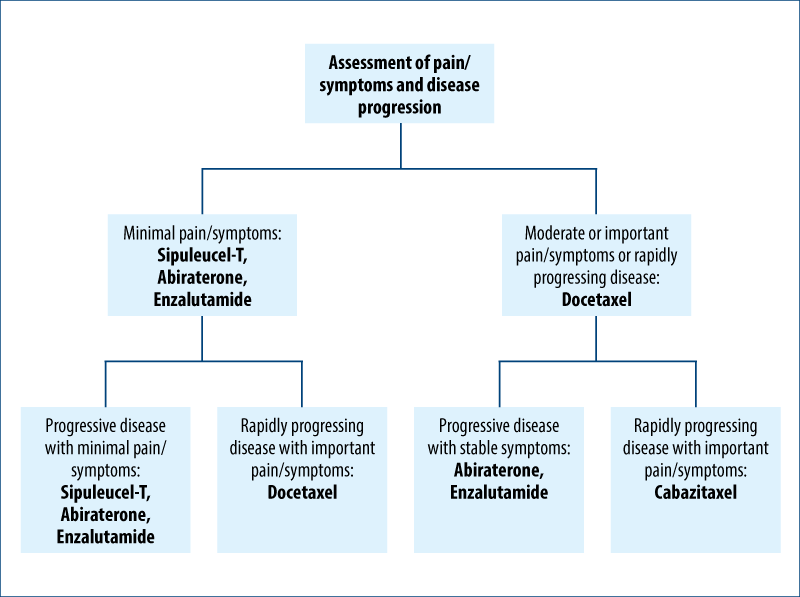
Recently, Cabazitaxel (Jeftana®), a semi-synthetic taxane, demonstrated survival benefits compared to Mitoxantrone, in patients with progressive disease following Docetaxel therapy (15.1 vs. 1.7 months).
Palliative CRPC treatment
Bone metastases therapy
PC patients have a high risk of developing bone metastases (MTS), which are associated with an increased incidence of adverse skeletal events (ASE) (from pathological fractures to spinal cord compression). Additionally, ASE risk increases due to anti-androgen therapy and corticosteroid therapy, which are chronically administered to these patients. For this reason, therapies aimed at preserving bone integrity are an essential part of CRPC management.
Bisphosphonates inhibit osteoclast activity to decrease bone turn-over. The only FDA-approved agent for bone MTS therapy in CRPC patients is zoledronate (4 mg i.v. every 3-4 weeks). Studies show a 25% reduction of ASE frequency in CRPC patients treated with zoledronate, with no other survival improvement. Denosumab is a monoclonal antibody that binds to RANKL and alters osteoclast function; although randomized trials showed no statistically significant difference in the frequency of side effects, disease progression or overall survival between denosumab and zoledronic acid, denosumab does not currently have FDA approval for treatment of bone MTS in CRPC.
Radiotherapy is a commonly used therapeutic option for painful bone MTS. Approximately 80% of patients describe pain improvement in the case of vertebral MTS irradiation. Most often, external irradiation of 30 Gy is used, in a single dose (same efficiency as multifractional regimes). Side effects include anaemia and fatigue, reversible most of the time. Radiotherapy must be associated with systemic control of the disease for optimal outcome of irradiated lesions.
Radiopharmaceuticals are a modern therapeutic option that has proved particularly effective in CRPC patients. Due to their preference for actively metabolic bone regions, they will be internalized in large quantities in the tumour and will induce breaks in the tumour DNA strands. The advantage of these agents lies in the fact that they can be administered in the case of generalized skeletal pain and multiple bone MTS. Currently, in the US, 3 radiopharmaceutical agents are approved: strontium 89, samarium lexidronam and radium-223 dichloride.
Bibliografie
2. Ahmadi H, Daneshmand S. Androgen deprivation therapy: evidence-based management of side effects. BJU Int 2013;111:543–548.
3. Antonarakis ES, Feng Z, Trock BJ, et al. The natural history of metastatic progression in men with prostate-specific antigen recurrence after radical prostatectomy: long-term follow-up. BJU Int 2012;109:32–39.
4. Attar RM, Takimoto CG, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clinical Cancer Research, vol. 15, no. 10, pp. 3251–3255, 2009.
5. Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 2008;26:4563–4571.
6. Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424–433.
7. Beltran H, Tomlins S, Aparicio A, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res 2014;20:2846–2850.
8. Bouchet BP, Galmarini CM. Cabazitaxel, a new taxane with favorable properties. Drugs Today (Barc) 2010;46:735–742.
9. Caubet JF, Tosteson TD, Dong EW, et al. Maximum androgen blockade in advanced prostate cancer: a meta-analysis of published randomized controlled trials using nonsteroidal antiandrogens. Urology 1997;49:71–78.
10. Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 2009;10:981–991.
11. Courtney KD, Taplin ME. The evolving paradigm of second-line hormonal therapy options for castration-resistant prostate cancer. Curr Opin Oncol 2012;24:272–277.
12. Crook JM, O’Callaghan CJ, Duncan G, et al. Intermittent androgen suppression for rising PSA level after radiotherapy. N Engl J Med 2012;367:895–903.
13. Danila DC, Morris MJ, de Bono JS, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol 2010;28:1496–1501.
14. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010;376:1147–1154.
15. DeVita Jr. VT (editor). DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology (Cancer Principles and Practice of Oncology). Wolters Kluver Health, 2014, Philadelphia
16. Dutt SS Gao AC. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncology, vol. 5, no. 9, pp. 1403–1413, 2009.
17. Falkmer U, Jarhult J, Wersall P, et al. A systematic overview of radiation therapy effects in skeletal metastases. Acta Oncol 2003;42:620–633.
18. Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 2011;377:813–822.
19. Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2012;13:983–992.
20. Gartrell BA, Saad F. Managing bone metastases and reducing skeletal related events in prostate cancer. Nat Rev Clin Oncol 2014;11:335–345.
21. Glass TR, Tangen CM, Crawford ED, et al. Metastatic carcinoma of the prostate: identifying prognostic groups using recursive partitioning. J Urol 2003;169:164–169.
22. Hanley DA, Adachi JD, Bell A, et al. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012;66:1139–1146.
23. Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nature Clinical Practice Urology, vol. 6, no. 2, pp. 76–85, 2009.
24. Holzbeierlein JM, Castle EP, Thrasher JB. Complications of androgen-deprivation therapy for prostate cancer. Clin Prostate Cancer 2003;2:147–152.
25. Hussain M, Tangen CM, Higano C, et al. Absolute prostate-specific antigen value after androgen deprivation is a strong independent predictor of survival in new metastatic prostate cancer: data from Southwest Oncology Group Trial 9346 (INT-0162). J Clin Oncol 2006;24:3984–3990.
26. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–422
27. Labrie F, Dupont A, Belanger A. Complete androgen blockade for the treatment of prostate cancer. Important Adv Oncol 1985:193–217.
28. Merrick GS, Butler WM, Wallner KE, et al. Androgen deprivation therapy does not impact cause-specific or overall survival in high-risk prostate cancer managed with brachytherapy and supplemental external beam. Int J Radiat Oncol Biol Phys 2007;68:34–40.
29. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447–4454.
30. O’Donnell A, Judson I, Dowsett M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer 2004;90:2317–2325.
31. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med 2013;369:213–223
32. Perry MC (editor). Perry’s the chemotherapy source book, fifth edition. Wolters Kluver Health, 2012, Philadelphia
33. Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513–1520
34. Porter AT et al. Results of a randomized phase-III trial to evaluate the efficacy of strontium-89 adjuvant to local field external beam irradiation in the management of endocrine resistant metastatic prostate cancer. Int J Radiat Oncol Biol Phys 1993; 25: 805–813.
35. Quilty P et al. A comparison of the palliative effects of strontium-89 and external beam radiotherapy in metastatic prostate cancer. Radiother Oncol 1994; 31: 33–40.
36. Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J 2013;19:43–49.
37. Ryan CJ, Molina A, Li J, et al. Serum androgens as prognostic biomarkers in castration-resistant prostate cancer: results from an analysis of a randomized phase III trial. J Clin Oncol 2013;31:2791–2798.
38. Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138–148.
39. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–1197
40. Scher HI, Morris MJ, Basch E, et al. End points and outcomes in castration-resistant prostate cancer: from clinical trials to clinical practice. J Clin Oncol 2011;29:3695–3704.
41. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol 2005;23:8253–8261.
42. Schulze H, Senge T. Influence of different types of antiandrogens on luteinizing hormone-releasing hormone analogue-induced testosterone surge in patients with metastatic carcinoma of the prostate. J Urol 1990;144:934–941.
43. Shahinian VB, Kuo YF, Freeman JL, et al. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 2005;352:154–164.
44. Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol 2004;22:1025–1033.
45. Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 2006;24:3089–3094.
46. Sprenger CC, Plymate SR. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm Cancer 2014;5:207–217.
47. Stephenson AJ, Scardino PT, Kattan MW, et al. Predicting the outcome of salvage radiation therapy for recurrent prostate cancer after radical prostatectomy. J Clin Oncol 2007;25:2035–2041.
48. Sweeney C, Chen YH, Carducci MA, et al. Impact on overall survival (OS) with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly metastatic prostate cancer (mPrCa): An ECOG-led phase III randomized trial. J Clin Oncol 2014;32:Abstr LBA2.
49. Tannock I, Gospodarowicz M, Meakin W, et al. Treatment of metastatic prostatic cancer with low-dose prednisone: evaluation of pain and quality of life as pragmatic indices of response. J Clin Oncol 1989;7:590–597
50. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502–1512.
51. Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol 1996;14:1756–1764.
52. Valicenti RK, Thompson I Jr, Albertsen P, et al. Adjuvant and salvage radiation therapy after prostatectomy: American Society for Radiation Oncology/American Urological Association guidelines. Int J Radiat Oncol Biol Phys 2013;86:822–828.
53. Zumsteg ZS, Zelefsky MJ. Short-term androgen deprivation therapy for patients with intermediate-risk prostate cancer undergoing dose-escalated radiotherapy: the standard of care? Lancet Oncol 2012;13:e259–e269.