Patologie rară cu debut atipic şi abordare multidisciplinară
Rare disease with atypical onset and multidisciplinary approach
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening syndrome developed due to uncontrolled hyperinflammation and tissue destruction. In most cases, it is a pediatric disorder, but it can also occur in adults. The clinical presentation and the laboratory findings are often nonspecific, therefore it is difficult to diagnose. Yet, an early diagnosis and treatment can significantly improve survival. Hence, the treatment should be initiated as soon as possible if one has a high suspicion of HLH, even though the patient does not meet yet all the criteria. We present the case of a patient admitted with nonspecific symptomatology, namely jaundice, fatigue and significant non-intentional weight loss. The initial laboratory data suggested cirrhosis. The attempt of discovering the cause of hepatic dysfunction was initially unsuccessful. We suspicioned HLH only based on the myelogram. At that point, the patient met the criteria for the diagnosis of HLH, and we started the specific treatment. However, the results were not satisfactory, and the patient died three months after the initial presentation.Keywords
hemophagocytic lymphohistiocytosiscirrhosisjaundiceanemiacorticotherapyRezumat
Limfohistiocitoza hemofagocitară (HLH) este un sindrom rar, ameninţător de viaţă, cauzat de o inflamaţie necontrolată şi de distrucţia ţesuturilor. În cele mai multe cazuri este o patologie pediatrică, dar poate fi întâlnită şi la adulţi. Prezentarea clinică şi analizele de laborator sunt de cele mai multe ori nespecifice, prin urmare este dificil de diagnosticat. Cu toate acestea, diagnosticul şi tratamentul precoce pot îmbunătăţi semnificativ supravieţuirea. Prin urmare, tratamentul trebuie iniţiat rapid dacă există o suspiciune înaltă pentru HLH, chiar dacă pacientul nu îndeplineşte încă toate criteriile necesare. Prezentăm cazul unei paciente cu simptomatologie nespecifică: icter, oboseală şi scădere ponderală neintenţionată semnificativă. Încercarea de a descoperi cauza disfuncţiei hepatice a fost iniţial nereuşită. S-a ridicat suspiciunea de HLH odată cu efectuarea medulogramei. În acel moment, pacienta a îndeplinit criteriile pentru diagnosticarea HLH şi s-a iniţiat tratamentul specific. Cu toate acestea, rezultatele nu au fost favorabile, iar pacienta a decedat la trei luni de la prezentarea iniţială.Cuvinte Cheie
limfohistiocitoză hemofagocitarăcirozăicteranemiecorticoterapieIntroduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare but life-threatening syndrome arising from a severe, uncontrolled hyperinflammatory reaction and tissue destruction(1). HLH is categorized as familial or sporadic. Familial HLH is an autosomal recessive disorder that appears in early childhood(2). Sporadic HLH is associated with a variety of underlying conditions, the most frequent being infections, malignancy, inflammatory and autoimmune diseases, immunosuppression, hematopoietic stem cells transplantation, organ transplantation and acquired immunodeficiency syndrome(1). The diagnosis of HLH is difficult due to the rarity of this syndrome and the nonspecific clinical and laboratory findings. However, in order to obtain a better outcome, the diagnosis and treatment must not be delayed(3). Even when the specific treatment is applied promptly, many patients die due to sepsis and/or multiorgan failure(3).
Case presentation
We present the case of a 57-year-old female admitted to the internal medicine department for jaundice, fatigue and significant non-intentional weight loss. Also, she described a recent episode of respiratory tract infection.
Her past medical history was positive only for vitiligo and arterial hypertension. She was not a smoker or an alcohol consumer. The physical exam was relevant for marked jaundice, hepatomegaly, splenomegaly and flank dullness.
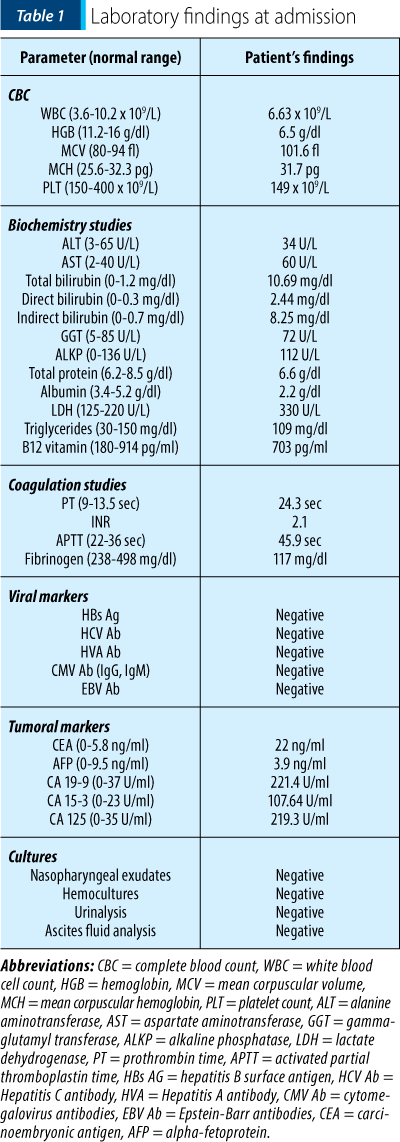
The laboratory data at admission (Table 1) showed severe macrocytic anemia, cholestasis, hypoalbuminemia and coagulation disturbances. The most tumoral markers were nonspecifically elevated. The Coombs tests, both direct and indirect, were negative and the vitamin B12 serum level was normal. The serum folates dosing was not available. The corrected reticulocytes count was 1.9%.
A series of imagistic investigations was performed, which revealed many signs of portal hypertension and its complications. The abdominal ultrasonography revealed a hypertrophic liver, confirmed the splenomegaly (160/65 mm) and the presence of ascites in high quantity. The computed tomography (CT) showed the presence of collateral circulation and permeabilization of the umbilical vein. The esophagogastroduodenoscopy revealed the presence of esophageal varices and portal gastropathy. None of the aforementioned investigations described formations suggestive of cancer.
An exploratory paracentesis was also performed. The ascites fluid exam showed sterile ascites, with a serum-ascites albumin gradient of 1.1 g/dl.
Considering the hepatomegaly, ascites, elevated bilirubin, hypoalbuminemia and coagulation anomalies, the diagnosis of cirrhosis was confirmed and graded as Child-Pugh class B. Up until now, the etiology of cirrhosis remained undiscovered. The patient was not an alcohol consumer and did not take hepatotoxic drugs. Also, no hepatotropic viruses were detected. To establish the cause of cirrhosis, we performed additional investigations. Anti-smooth muscle antibodies, anti-nuclear antibodies and anti-mitochondrial antibodies were negative, therefore we excluded autoimmune hepatitis as the cause of cirrhosis. The iron and copper metabolisms were studied. The laboratory findings showed increased ferritin, although the patient did not receive iron supplements or transfusions, and a low level of ceruloplasmin. The ophthalmologist raised the suspicion of the presence of Kayser-Fleischer ring. We performed an abdominal MRI (magnetic resonance imaging) that did not reveal any hypodensities suggestive of hepatocytic iron or copper overload. The MRI findings, together with the absence of ATP7b (ATPase Copper Transportation Beta) gene excluded the diagnosis of Wilson’s disease.
We performed a bone marrow aspirate and studied the cytology by optic microscopy. The results showed hypercellularity due to increased erythropoiesis, increased medullary hemosiderin, 81% sideroblasts, 1% ringed sideroblasts, and hemophagocytosis. The latter raised the suspicion of hemophagocytic lymphohistiocytosis.
The patient received specific treatment with corticosteroids and supportive treatment – RBC (red blood cells) transfusions, cryoprecipitate, fresh frozen plasma and broad-spectrum antibiotics. Due to her significant deterioration of organ function, immunosuppressive drugs could not be administered (such as etoposide).
Despite treatment, the clinical and biological status of the patient deteriorated. She developed extensive subcutaneous hematomas, and the jaundice became more severe. Even though she received RBC transfusions, the hemoglobin remained low. She gradually developed severe thrombocytopenia (30 x 109/L), hyponatremia, hyperpotassemia, nitrogen retention, the total bilirubin increased to 30.97 mg/dl, and the coagulation alteration became more significant, with a spontaneous INR of 3.16.
Three months after her admission, the patient suffered a cardiac arrest. She was successfully resuscitated, but maintained a severe comatose state (3 points on Glasgow Coma Scale). Afterwards, the patient was supervised in the intensive care unit. She died ten days after the resuscitated cardiac arrest.
Discussion
HLH is a rare syndrome that lacks specificity regarding the clinical signs and symptoms, and also in any gold standard diagnostic test. Therefore, establishing the diagnosis of HLH is particularly difficult. The current accepted criteria are the HLH-2004 criteria(4). Five out of the following eight findings must be met: fever ≥38.5ºC, splenomegaly, peripheral blood cytopenia (hemoglobin <9 g/dl, platelets <100 x 109/L, absolute neutrophil count <1 x 109/L), hypertriglyceridemia (fasting triglycerides >265 mg/dl) and/or hypofibrinogenemia (fibrinogen <150 mg/dl), hemophagocytosis in bone marrow, spleen, lymph node, or liver; low or absent NK cell activity, ferritin >500 ng/ml, and elevated soluble CD25(4). In our case, the patient met five criteria, namely splenomegaly, severe cytopenia, hypofibrinogenemia, elevated ferritin and hemophagocytosis in the bone marrow. The measurement of NK cell activity and soluble CD25 were not available.
The most common trigger for both familial and sporadic cases is immune activation from an infectious disease(1). Our patient described a recent episode of respiratory tract infection. While admitted, she was not febrile and showed no other signs of infection. The tests for viral markers were all negative. Another common trigger is malignancy(1). Our patient had mostly nonspecific slight elevations of tumor markers but without any cancer-suggestive markers in imaging studies. However, we cannot exclude the possibility of a malignancy.
The prompt treatment is critical in order to obtain a favorable outcome. The delays in the administration of the appropriate treatment can lead to shorter survival. Therefore, the treatment should be given when we have a high suspicion, even though the patient meets less than five criteria(5). The guidelines recommend the treatment of the underlying disease, corticotherapy and etoposide, with or without intrathecal therapy, along with supportive care(6). In our case, the patient met five criteria out of eight. The hemophagocytosis discovered in the bone marrow was crucial in establishing the diagnosis of HLH, given the nonspecific clinical evolution. The patient received promptly specific treatment with corticosteroids.
The adult HLH has an unpredictable course of evolution and usually unfavorable. The survival of untreated patients with HLH is approximately two months(6). Despite specific and supportive treatment, our patient died three months after her admission.
Conclusions
HLH is a rare, complex and severe syndrome. It is characterized by nonspecific clinical and paraclinical findings and by evolution towards multiple organ dysfunction syndrome. The delay in diagnosis and treatment is translated into a shortened survival. Although our patient received specific treatment for HLH, it had little impact since the nonspecific presentation made the diagnosis difficult and delayed the treatment.
Conflicts of interests: The authors declare no conflict of interests.
Bibliografie
- Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol. Am Soc Hematol Educ Program. 2013;2013:605-611. DOI: 10.1182/asheducation-2013.1.605
- Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lyphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163(3):271-283. DOI: 10.1111/j.1365-2249.2010.04302.x
- Yoon JH, Park SS, Jeon YW, Lee SE, Cho BS, Eom KS, Kim YJ, Lee S, Min CK, Cho SG, Lee JW. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematol. 2019; 104(2): 269-276. DOI: 10.3324/haematol.2018.198655
- Henter JI, Horne A, Arico M, Egeler RA, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. DOI: 10.1002/pbc.21039
- Khan HH, Ansar I, Kontos N, Kumar S, Lyons H. Report of a Fatal Case of Hemophagocytic Lymphohistiocytosis Syndrome and a Review of the Literature. Cureus. 2020;12(12):e12049. DOI: 10.7759/cureus.12049
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052. DOI: 10.1182/blood-2011-03-278127

Imagistica trombozei venoase tumorale din carcinomul hepatocelular – esenţialul
Mihai Pomohaci, Miruna Ispas, Ioana G. Lupescu
Obiectiv. Scopul acestei lucrări este de a analiza prevalenţa trombozei tumorale (la nivelul venei porte, venelor hepatice şi al venei cave inferioare) la pacienţii cu carcinom hepatocelular (CHC), în corelaţie c...
Modificări hematologice în ciroza hepatică – o provocare în practica clinică
Minodora Onisâi, Ana Maria Vlădăreanu, Iuliana Iordan, Cristina Enache, Ovidiu Calapod, Andreea Marin, Carmen Georgeta Fierbinţeanu-Braticevici, Tribus Laura Carina
Ciroza reprezintă stadiul final de evoluţie în multiple patologii hepatice. Pacienţii cu ciroză hepatică prezintă anomalii pr...
Regimul FOLFIRINOX în tratamentul adjuvant şi metastatic al cancerului pancreatic în era oncologiei de precizie
Anda Crişan, Liliana Eleonora Semenescu, Ciprian Camil Mireştean, Adina Mitrea, Irina Roxana Iancu, Dragoş Petru Teodor Iancu
Cancerul pancreatic rămâne o boală agresivă, cu un prognostic nefavorabil, deşi s-au făcut progrese în ultimii ani în ceea ce pri...
Cardiomiopatia cirotică – factor de prognostic negativ în ciroză
Andreea Maria Marin, Ovidiu Calapod, Gabriela Anca Angelescu, Corina Costache, Ruxandra Sfeatcu, Tribus Laura Carina
Cardiomiopatia cirotică reprezintă o formă de afecţiune cardiacă ce apare la pacienţii cu ciroză, în absenţa unei boli cardiace cunoscute. ...
Legătura complexă dintre ficatul gras nonalcoolic şi diabetul zaharat de tip 2
Tribus Laura Carina, Andreea Maria Marin, Ovidiu Calapod
Boala ficatului gras nonalcoolic (FGNA) reprezintă o problemă de sănătate publică la nivel mondial, fiind o importantă sursă de morbiditate şi mortalitate în rândul populaţiei. Prevalenţa acestei boli este într-o continu...