Un caz rar de asociere a unui cancer renal, mamar şi colorectal la o pacientă tânără
A rare case of associated renal, breast and colorectal cancer in a young patient
Abstract
We are presenting the case of a young woman with associated renal carcinoma, bilateral breast cancer, metastatic colorectal cancer and benign tumor of the ovary. The patient’s family history includes ovarian clear cell carcinoma, colorectal cancer, prostate carcinoma, breast cancer, melanoma and pulmonary carcinoma. First, she had surgery at the age of 28 years old for the ovarian mass, followed by the diagnosis of renal clear cell carcinoma at 42 years old, breast cancer at 47, colorectal cancer and renal cell carcinoma recurrence at 49 years old, and a new breast cancer at 50 years old. We tried to analyze the history and to include the patient in one of the most common familial cancer syndromes, taking into account that she has also a personal and a familial history of multiple neoplasms.Keywords
renal clear cell carcinomagenetic syndromemultiple primary neoplasmsbreast cancerRezumat
Prezentăm cazul unei femei tinere cu asociere de carcinom renal, cancer mamar bilateral, cancer colorectal metastatic şi tumoră benignă a ovarului. Istoricul familial al pacientei include carcinom ovarian cu celule clare, cancer colorectal, carcinom de prostată, cancer de sân, melanom şi carcinom pulmonar. Pacienta a fost operată la vârsta de 28 de ani pentru masă ovariană, urmată de diagnosticarea carcinomului renal cu celule clare la 42 de ani, a cancerului de sân la 47 de ani, a cancerului colorectal şi a recidivei carcinomului renal la 49 de ani şi a unui nou cancer de sân la 50 de ani. Am încercat să analizăm istoricul şi să includem pacienta într-unul dintre cele mai frecvente sindroame familiale ce asociază mai multe neoplazii, având în vedere istoricul personal şi familial de neoplasme multiple.Cuvinte Cheie
carcinom renal cu celule claresindrom geneticneoplasme primare multiplecancer de sânIntroduction
Diagnosing a cancer at a young age, multifocal, with multiple unrelated primary neoplasms, along with a family history of neoplastic disease and congenital malformations makes the diagnosis of a hereditary cancer a high possibility. Only about 5% to 10% of all cancers result directly from inherited gene mutations.
We present the case of a woman who was diagnosed between the ages of 28 and 50 years old with benign ovarian tumor, renal carcinoma, bilateral breast cancer, metastatic colonic cancer and Hashimoto disease.
Case report
The patient I.C. is a 50-year-old Caucasian woman with a medical history of multiple neoplasms and benign tumors, arterial hypertension and Hashimoto thyroiditis.
First, she was diagnosed in 1999, at 28 years old, with a papillary ovarian benign tumor for which right anexectomy was practiced, with good postoperatory outcome. There was no other investigation performed.
In 2013, she presented at her general practitioner for hematuria. After multiple investigations, an abdomino-pelvic scan revealed the presence of a mass measuring 38/35 mm developed in the right kidney. In April 2013, the patient suffered a right nephrectomy and the hystopathology demonstrated that the 4-cm mass was a clear cell renal carcinoma, moderately differentiated, with minimum invasion of the renal capsule. Later in the same year, the patient started to complain of extreme fatigue and muscular pain. The investigations performed revealed abnormal thyroid hormone levels and the presence of antithyroid peroxidase antibodies. In December 2013, the patient underwent surgery and a thyroidectomy was performed. The histopathology analysis confirmed the diagnosis of Hashimoto disease.
In March 2018, at a routine medical exam, a breast mass was found. The histopathology and immunohistochemistry performed from the tumor sample tissue revealed a type B mucinous carcinoma, moderately differentiated, with extensive areas of in situ ductal carcinoma, ER 90%, PgR 80%, CerbB2 negative (1+) and a Ki67 of 25%. The patient underwent neoadjuvant treatment with chemotherapy EC90 followed by docetaxel 90 mg/m2 and modified radical mastectomy. After the surgical intervention, she started hormonotherapy with tamoxifenum 20 mg/day and goserelinum 3.6 mg/28 days.
At an imagistic follow-up, in February 2020, a tumoral nodule was discovered in the right nephrectomy area. The surgeon decided to operate her and the histopathological exam indicated a clear cell renal carcinoma metastasis in the right adrenal gland, therefore the medical oncologist decided to start the therapy with sunitinibum.
No other medical condition was diagnosed until March 2021, when the patient experienced diarheea alternated with constipation. The gastroenterologist found at colonoscopy a stenosis at 40 cm of the anal orifice. The histopathology exam from the collected tissue sample came back positive for intestinal adenocarcinoma, moderately differentiated, with microsatellite stability and KRAS gene mutation. Moreover, the CT scan performed in April 2021 revealed multiple hepatic masses, with the aspect of secondary determinations.
The patient adressed to our clinic in April 2021. Our multidisciplinary team decided to operate her. The surgical team performed a left hemicolectomy and metastasectomy with R0 resection. The final stage was ypT3 ypN2b, with the confirmation of the previous histopathology and immunohistochemistry profile. In May 2021, she started the adjuvancy with chemotherapy – FOLFOX6 protocol. During the precycle clinical examination, in August 2021, we observed the presence of a suspect mass in the restant breast. A mammography was performed which showed a suspect lesion, with a BI-RADS score of 5. In the absence of any clinical or imagistic sign of adenopathy, an excisional biopsy was performed that revealed the presence of a ductal carcinoma, with ER 90%, PgR 90%, HER2 expression negative and Ki67 of 1-2%. The patient will follow hormonotherapy with anastrozole 1 mg/day.
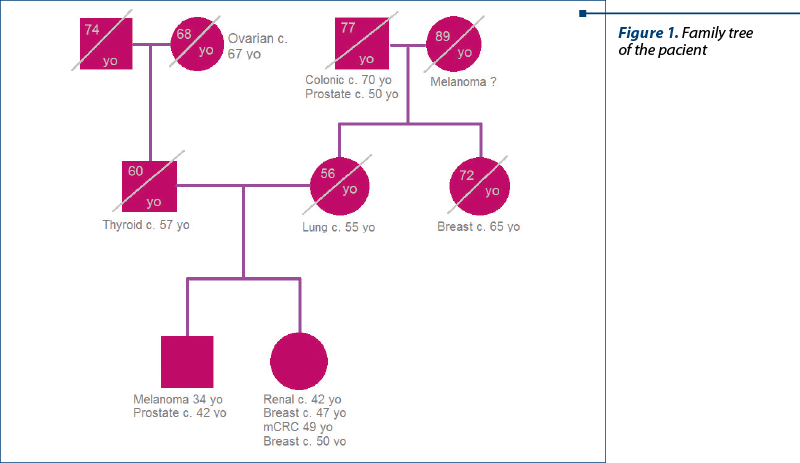
The patient refused to see a medical genetician, as she does not have any children. From her family history, we know that her father suffered from a thyroidian carcinoma (diagnosed at 57 years old), her aunt (maternal side) had a breast cancer, the paternal grandmother died from an ovarian cancer (68 years old), her maternal grandfather suffered from a prostate cancer and died from a colonic cancer (77 years old), and her older brother was diagnosed with melanoma at 34 years old and with prostate cancer at 42 years old, being currently free of disease.
Discussion
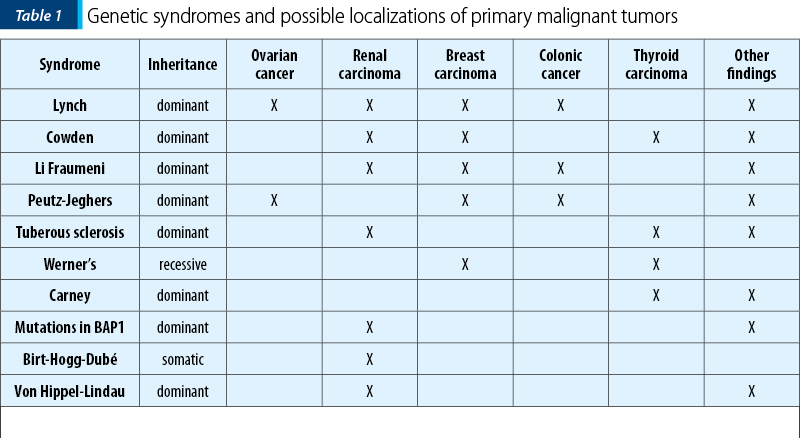
We report a case with several cancers that could not fall into any of the main family cancer syndromes. Family history is significant for cancers of the colon, thyroid, prostate, lung, melanoma, breast and ovary, in addition to the early diagnosis of a benign lesion of the ovary at the age of 28 years old.
The possibility of adenomatous polyposis coli (APC) syndrome is ruled out in this case because the polyposis is not constituted and no analysis of the APC gene mutation has been performed. In addition, the multiple cancers diagnosed did not fit with this syndrome, when compared to what is published about APC syndrome.
Hereditary nonpolyposis colon cancer (HNPCC) is another syndrome with mutations reported in the repair mismatch gene (MMR) complexes that include several mutations such as hMLH1 at 3p21.3, hMSH2 at 2p22-p21, hPMS1 at 2q31-q33, hPMS2 at 7p22 and hMSH6 at 2p16. The diagnosis is based on a family history of colon cancer diagnosed before the age of 50, without a detected family adenomatous polyposis. Associated malignancies are colorectal, ovarian, endometrial, urothelial, gastric, pancreatic, hepatobiliary carcinoma, glioblastoma and breast cancer. The associated benign neoplasms are colonic adenomas, epithelioma, keratoacanthomas and sebaceous adenomas(1,2).
Cowden syndrome is reported with mutations in the PTEN gene at 10q23. The clinical diagnosis is based on epidermal lesions, intestinal hamartomas, macrocephaly and cerebellar glial mass that leads to impaired gait and convulsions (Lhermitte-Duclos disease). The associated malignancies are colon, endometrial, kidney, breast, lung, thyroid, ovarian and Merkel cell carcinomas, melanoma and retinal glioma. The associated benign lesions are cutaneous warts of the face and limbs, hyperkeratosis of the gums and oral mucosa, facial trichylemomas, hyperkeratosis of the hands and feet, hamartomatous polyps, adenomas, lipomas and hemangiomas(3,4).
In the literature, von Hippel-Lindau syndrome (VHL) and mutations in VHL have been associated with clear cell carcinoma of the kidney. Von Hippel-Lindau syndrome is also characterized by CNS hemangioblastoma (brain, spine, retina), pancreatic and renal cysts, pheochromocytoma, endolymphatic sac tumors and neuroendocrine tumors of the pancreas(5).
Birt-Hogg-Dubé disease (BHD) is an autosomal dominant syndrome with a high neoplasm susceptibility. It is characterized by the development of fibrofolliculomas, lung cysts, spontaneous pneumothorax and renal cancer and is due to mutations in folliculin (FLCN)(6). The dermatologic features and lung disease are the most common presenting features; BHD is underdiagnosed due to its variable, often mild, presentation. A wide spectrum of renal cancers has been observed in patients with this syndrome(7,8).
BAP1 mutations (BRCA-associated protein 1) were identified through somatic sequencing of renal tumors. Studies found that BAP1 mutations can lead to familial clear cell carcinoma of the kidney and to uveal/cutaneous melanoma and mesothelioma(8).
Conclusions
Given the fact that the level of knowledge and the possibilities of diagnosis have increased exponentially in recent decades, we need to know as deeply as possible about genetic syndromes.
The interdisciplinary collaboration with medical geneticists must become the standard of care in order to be able to manage these patients as accurately as possible.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997 Dec 3;89(23):1758-62.
-
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995 Mar 30;332(13):839-47.
-
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997 May;16(1):64-7.
-
Brownstein MH, Mehregan AH, Bikowski JB, Lupulescu A, Patterson JC. The dermatopathology of Cowden’s syndrome. Br J Dermatol. 1979 Jun;100(6):667-73.
-
Tickoo SK, de Peralta-Venturina MN, Harik LR, Worcester HD, Salama ME, Young AN, Moch H, Amin MB. Spectrum of epithelial neoplasms in end-stage renal disease: An experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol. 2006;30:141–153.
-
Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, Sharma N, Walther M, Munroe D, Hill R, Maher E, Greenberg C, Lerman MI, Linehan WM, Zbar B, Schmidt LS. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–164.
-
Pavlovich CP, Grubb RL 3rd, Hurley K, Glenn GM, Toro J, Schmidt LS, Torres-Cabala C, Merino MJ, Zbar B, Choyke P, Walther MM, Linehan WM. Evaluation and management of renal tumors in the Birt-HoggDubé syndrome. J Urol. 2005;173:1482–1486.
-
Haas NB, Nathanson KL. Hereditary kidney cancer syndromes. Advances in Chronic Kidney Disease. 2014;21(1):81-90.

Hipereozinofilia în limfoamele maligne non-hodgkiniene T-periferice – mai mult decât o modificare hematologică
Diana Emanuela Bonea, Cristina Mambet, Mihaela Găman, Iuliana Iordan, Andreea Neculcea, Alina Grigoroiu, Patricia Pîrvan, Dragoş-Claudiu Popescu, Onofrei Alexandru , Cristina Enache, Stejara Nicoleta Mihai, Ferea Roxana Cătălina , Alexandra Maria Baciu, Ana Maria Vlădăreanu
Eozinofilia este o modificare hematologică frecventă, asociată cu neoplaziile hematologice, cu tumorile solide sau cu boli autoim...
Debut cu tromboze extensive în trombocitemia esenţială triplu negativă – dificultăţi de diagnostic. Prezentare de caz
Tiberiu Sultan, Anca Nicolescu, Iuliana Iordan, Cristina Mambet, Cristina Enache, Stejara Nicoleta Mihai, Raluca Nistor, Paul Findrihan, Diana Cisleanu, , Ana Maria Vlădăreanu
Trombocitemia esenţială (TE) este un neoplasm mieloid cronic caracterizat prin trombocitoză persistentă nonreactivă (trombocit...
Aspecte IRM ale leziunilor PI-RADS 5 – experienţa unui centru terţiar
Sandra O. Jurcă, Gabriel Gluck, Ioana G. Lupescu
Cancerul de prostată este una dintre cele mai frecvente afecţiuni maligne în rândul bărbaţilor, pentru care se depun eforturi sporite, cu scopul de a îmbunătăţi rata de diagnosticare timpurie şi de a preveni cazu...
Intratumoral heterogeneity in breast cancer – a real challenge for diagnosis and treatment
Marius Pirciu, Elena Avram, Iolanda Georgiana Augustin, Daniela Zob, Andreea Lăzescu
Prezentăm cazul unei paciente cu neoplasm mamar la care, în evoluţia bolii, prin biopsii multiple, s-a remarcat o pronunţată heterogenitate intratumorală din punctul de vedere al constelaţie...
Fighting disease at 83 years old: SARS-CoV-2 survival and response to anti-PD-1 therapy in PD-L1 negative adenocarcinoma of the lung – case report
Maria Nedelcu, Adelina Silvana Dragomir, Elena Adriana Mateianu, Dana Lucia Stănculeanu
În timpul pandemiei de COVID-19, supravieţuirea totală medie a pacienţilor cu cancer pulmonar a scăzut de la 7,9 luni la 6,7 lu...