Prevalenţa mutaţiilor 35delG şi W24X la copiii cu hipoacuzie congenitală non-sindromică
Prevalence of 35delG and W24X mutations in non-syndromic congenital hearing loss
Abstract
During the last decade, due to outstanding progress in understanding the molecular bases of sensoneural hearing loss, it became clear that 50% to 80% of all such afflictions result from mutations in a single gene, GJB2, which encodes the protein Connexin 26. One mutation of this gene, the 35delG allele, is particulary common in white populations. Studying this matter within the Romanian population is still in its early stages. In order to, at least partialy clarify this problem, we searched for the two most common variations of this gene, 35delG and W24X, in children with positive diagnosis of bilateral severe - profound sensoneural hearing loss.Keywords
Connexin 26GJB2W24Xcongenital nonsyndromic hearing lossgenesRezumat
Datorită progreselor deosebite înregistrare în ultima decadă în ceea ce priveşte înţelegerea bazelor moleculare ale apariţiei hipoacuziei neuro-senzoriale, s-a putut stabili că între 50% şi 80% din aceste hipoacuzii au drept cauză mutaţia unei singure gene, GJB2 care codifică o proteină cunoscută sub numele de Connexina 26. Mutaţia 35delG este descrisă ca fiind cea mai frecvent întâlnită la populaţia de tip caucazian. Studiul prevalenţei acestei mutaţii în populaţia generală a României se găseşte încă în perioada de pionierat. Pentru a încerca să elucidăm măcar parţial această temă, am căutat să evidenţiem cele mai frecvente două mutaţii ale genei, 35delG şi W24X, la copii diagnosticaţi cert cu hipoacuzie neuro-senzorială severă sau profundă bilaterală.Cuvinte Cheie
Connexina 26GJB2W24Xhipoacuzie congenitală non-sindromicăgeneIntroducere
Conform datelor raportate de Organizaţia Mondială a Sănătăţii, actualmente în populaţia globului se înregistrează peste 250 de milioane de persoane ce suferă de afecţiuni ale auzului de diverse etiologii, ceea ce reprezintă 4,2% din populaţia globului(4,5,19,21). Hipoacuzia congenitală este o afecţiune relativ frecventă, cu o incidenţă ce variază conform diverselor surse din literatură de la 1-3/1000 nou-născuţi(26) la 1/500 nou-născuţi(33). Deşi heterogene din punct de vedere etiologic, cel puţin 50% din hipoacuziile cu debut precoce au cauze genetice, iar dintre acestea, majoritatea sunt cel mai probabil autozomal recesive (75-80%)(33) şi non-sindromice (70%)(26,33,13,24). Din cauza heterogenităţii etiologice a hipoacuziei, evaluarea şi sfatul genetic ridică probleme, în special în cazul copiilor cu vârste foarte mici. Cu toate acestea, descoperirile recente indică mutaţiile genelor GJB2 şi GJB6 de pe cromozomul 13q11-q12 drept responsabile de peste 50% din formele de hipoacuzie congenitală non-sindromică autozomal recesivă în anumite grupuri populaţionale. Acest fapt uşurează în mare măsură diagnosticul genetic şi oferă familiilor şanse sporite de a obţine informaţii cu privire la o astfel de afectare genetică.
Genele GJB2 şi GJB6 codează proteinele membranare Connexina 26 (Cx26) şi Connexina 30 (Cx30) ce alcătuiesc canalele intercelulare heteromerice numite gap-junction, structuri cu rol în homeostazia cohleară, ce asigură fluxul de ioni de K+ de la celulele ciliate interne şi externe la celulele de susţinere, apoi la fibrocitele ligamentului spiral şi ale limbului spiral şi înapoi în endolimfă. Lista de variante allelice pentru GJB2 cuprinde peste 100, în special pentru formele autozomal recesive. Deleţia unei singure guanine (35delG) este responsabilă pentru 50% din cazurile de hipoacuzie neurosenzorială non-sindromică în populaţia Europei, Americii de Nord şi Asiei(22,25,38). Deleţia comună 342-Kb a GJB6 (numită GJB6-D13S1830) apare la până în 20% din hipoacuzicii din S.U.A. şi poate fi responsabilă de aproximativ 10% din toate allelele DFNB1, având o rază de acţiune foarte largă, bazată pe originea etnică şi adesea în asociere digenică cu varianta 35delG/GJB2(11,14,25).
Studii foarte recente au formulat şi o altă ipoteză conform căreia mutaţiile genei GJB6 nu ar avea nici o contribuţie în apariţia hipoacuziei şi că proteina Connexina 30 nu ar face decât să moduleze acţiunea Connexinei 26, iar hipoacuzia legată de cele trei mutaţii dominante cunoscute ale GJB6 ar reflecta alterări funcţionale ale canalelor heteromerice ce conţin Cx26 normală şi Cx30 modificată. Astfel, la pacienţii surzi care prezintă o deleţie a GJB6, cauza hipoacuziei ar fi un defect al Connexinei 26 modulat de mutaţia genei GJB6(6).
O afectare genetică aduce după sine şi consecinţe sociale şi psihologice asupra familiei (grija pentru starea de sănătate a copilului, sentimentul de stigmatizare, sentimentul de vinovăţie cu privire la transmiterea unei atare afecţiuni către copil) care pot afecta capacitatea părinţilor de a se adapta situaţiei(17). După naşterea unui copil surd, părinţii nu cunosc de obicei etiologia afecţiunii iar majoritatea nu se aşteaptă la o cauză genetică, în special când nu există un istoric familial sugestiv(34).
Material şi metodă
Studiul a fost efectuat pe un lot de 30 de copii (18 fete şi 12 băieţi) cu vârste cuprinse între 2 şi 10 ani (vârsta medie: 4,62 ani), proveniţi din 29 de familii fără grad de rudenie între ele, diagnosticaţi cu hipoacuzie severă sau profundă bilaterală congenitală în cadrul Secţiei O.R.L. a Spitalului Clinic de Urgenţă pentru copii „Marie Sklodowska Curie” în perioada octombrie 2014 - aprilie 2015. După elucidarea istoricului familial şi personal al pacientului, bazat pe un interviu personalizat, s-a efectuat examenul clinic O.R.L. complet. Acesta a avut drept scop excluderea formelor sindromice de hipoacuzie congenitală sau a altor patologii asociate. Interviul personalizat a avut drept scop obţinerea de informaţii esenţiale cu privire la istoricul sarcinii (starea de sănătate a mamei, expunerea la poluanţi, utilizarea de medicamente, în special ototoxice), al naşterii (prematuritatea, hipoaxia, hiperbilirubinemia, traumatismele craniene, perioada petrecută în secţia de Terapie Intensivă Neonatală etc.) şi evoluţia hipoacuziei (vârsta la care a fost observată).
Diagnosticul audiologic a fost stabilit în funcţie de vârsta copilului (după excluderea unei patologii de ureche medie), fie pe baza testării prin otoemisiuni acustice (OAE) şi potenţiale evocate de trunchi cerebral (BERA şi ASSR) pentru copiii cu vârsta sub 4 ani (cu respectarea normelor impuse de ghidurile internaţionale în vigoare)(2), fie prin audiogramă tonală liminară pentru copiii cu vârsta peste 4 ani. Cazurile de hipoacuzie dobândită sau formele sindromice de hipoacuzie congenitală nu au fost incluse în studiu. În studiu nu au fost incluşi pacienţi cu istoric familial sugestiv pentru hipoacuzie dar au fost păstraţi pacienţii ce prezentau în antecedente factori de risc pentru apariţia hipoacuziei congenitale (tratamente ototoxice, hipoxia la naştere, prematuritatea etc.) deoarece s-a constatat că prezenţa acestor factori nu exclude automat prezenţa mutaţiilor genetice. Pacienţii incluşi în studiu nu au beneficiat de screening auditiv neo-natal chiar dacă unii dintre ei aveau factori de risc cerţi pentru apariţia hipoacuziei. Vârsta medie de diagnosticare a hipoacuziei a fost de 2,7 ani.
După obţinerea consimţământului informat al părinţilor s-au prelevat probe de sânge periferic pentru efectuarea de teste moleculare şi stabilirea unei posibile cauze genetice a hipoacuziei. Prelevarea s-a efectuat în eprubete cu EDTA. ADN-ul a fost extras din sângele periferic prin cultivarea in vitro a limfocitelor. Culturile au fost oprite în metafază cu colchicină. Analiza moleculară s-a realizat prin amplificarea directă a ADN-ului din genom folosind primeri specifici prin metoda PCR. Tehnicile folosite au fost AS-PCR şi multiplex PCR pentru analiza mutaţiilor 35delG şi W24X ale genei GJB2.
Rezultate
Întrucât testarea genetică este o analiză costisitoare iar mijloacele financiare alocate studiului limitate, s-a efectuat o selecţie a pacienţilor. Iniţial am evaluat un lot extins de 52 de pacienţi copii, cu hipoacuzie neuro-senzorială severă sau profundă bilaterală. Au fost selectaţi 30 dintre aceştia (57% din cazuri), copii care nu asociau alte probleme de sănătate (forme non-sindromice) şi care proveneau din părinţi cu auz normal. Aceşti 30 de subiecţi au fost supuşi unui screening molecular pentru determinarea mutaţiilor 35delG şi W24X ale genei GJB2.
Pentru mutaţia 35delG, 26,6% din pacienţi (8/30) se încadrează în grupul homozigot, cunoscut drept genotip Δ/Δ (litera Δ care precede numele unei gene semnifică faptul că acea genă prezintă o deleţie cromozonială, iar litera N reprezintă lipsa deleţiei), iar 6,66% din pacienţi (2/30) aparţin grupului heterozigot digenic 35delG/W24X. Nu a fost descoperit nici un caz de pacient ce aparţine grupului heterozigot, genotip Δ/N. Mutaţia unică W24X s-a regăsit sub formă homozigotă la 6,66% din pacienţi (2/30) şi în formă heterozigotă la 3,33% din pacienţi (1/30). Rezultatele sunt prezentate în tabelul 1.
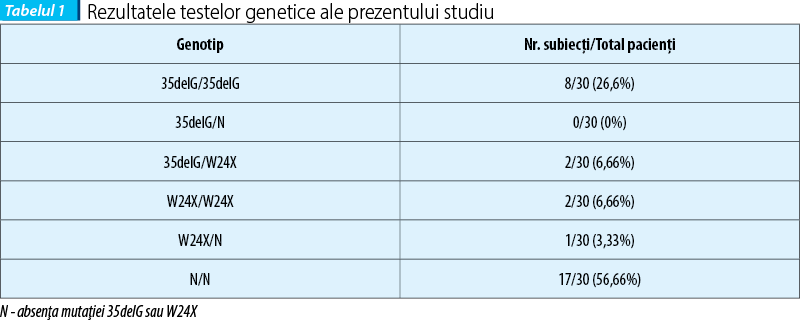
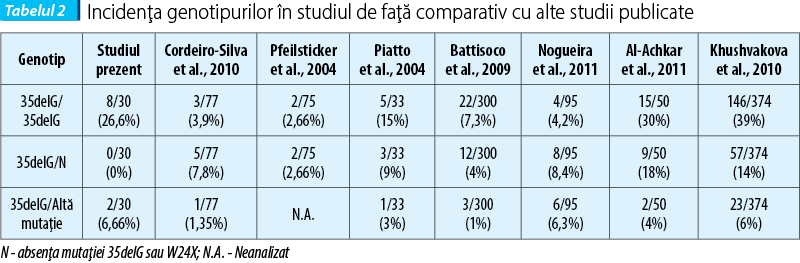
Incidenţa de apariţie a allelei mutante 35delG în lotul de studiat a fost de 33%, în concordanţă cu studiile publicate anterior în literatura, iar a allelei W24X de 16% (tabelul 3).
Discuţii
Hipoacuzia neuro-senzorială reprezintă una dintre cele mai frecvent întâlnite dizabilităţi ale omului iar factorul genetic joacă un rol principal în cercetarea şi diagnosticul acesteia. Până în prezent au fost identificaţi peste 130 de loci ai hipoacuziei non-sindromice la om şi se estimează că peste 100 de mutaţii ale genelor connexinei 26 ar fi implicate(1). Diagnosticul genetic al hipoacuziei neuro-senzoriale congenitale non-sindromice este deosebit de dificil din cauza varietăţii de mutaţii descrise drept răspunzătoare de apariţia ei. Ca factor adiţional, predominanţa unei anumite gene implicate variază semnificativ cu tipul de populaţie.
Mutaţia 35delG a genei GJB2 rămâne principala cauză de apariţie a surdităţii cu substrat genetic în populaţia de tip caucazian, cu o incidenţă a purtătorilor care variază de la 1/35 în populaţia sud-europeană la 1/79 la cea nord-europeană(10,12) şi o incidenţă maximă în ţările din jurul Mediteranei(1,8,18). Mutaţia 35delG este, de asemenea, una dintre cele mai frecvente mutaţii cu potenţial patogen la om, cu o frecvenţă a purtătorilor asemănătoare cu mutaţia deltaF508 a fibrozei chistice (CFTR)(10,37). Ea poate fi regăsită atât ca homozigot cât şi ca heterozigot. De asemenea, se poate asocia cu alte mutaţii ale genei GJB2 dar şi cu mutaţii ale genei GJB6 precum D13S1830 care este a doua mutaţie ca frecvenţă în apariţia hipoacuziei non-sindromice în populaţia europeană. La anumite grupuri populaţionale precum japonezii, chinezii, evreii aşkenazi sau populaţiile de origine rromă, mutaţia 35delG este rară, locul ei în cadrul etiologiei genetice a hipoacuziei congenitale fiind preluat de alte mutaţii precum 235delC, 167delT sau W24X(15).
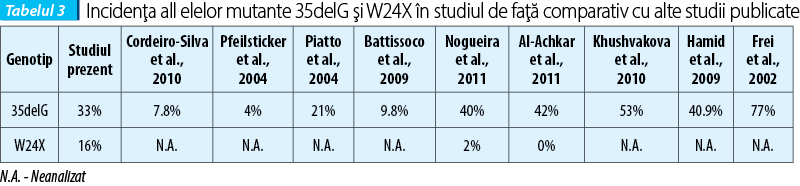
Studiul de faţă evidenţiază incidenţa de apariţie a mutaţiei 35delG şi secundar a mutaţiei W24X în populaţia afectată de hipoacuzie congenitală. Cu toate că s-a limitat la cazurile prezentate într-o singură clinică şi la un număr relativ mic de probe prelevate, studiul a relevat că screening-ul a două etiologii comune precum mutaţiile 35delG şi W24X şi selecţia riguroasă a cazurilor, pot evidenţia prezenţa factorului genetic la aproape 50% dintre pacienţii cu hipoacuzie neuro-senzorială congenitală severă sau profundă bilaterală de cauză necunoscută. Incidenţa 35delG a fost de 26,6%, în concordanţă cu studii asemănătoare ce raportează, în funcţie de populaţia studiată, 13,6% în Iordania, 14% în Palestina(1,32) 16% în Egipt(1,23) 5,66% în Iran(1,22) 94% în Liban(1,16) şi 60-80% în populaţiile europene(1,35) (tabelul 2) în vreme ce frecvenţa relativă a tuturor allelelor mutante 35delG a fost de 33%, de asemenea în în concordanţă cu studiile publicate anterior în literatura (tabelul 3). Cercetările efectuate în Orientul Mijlociu au determinat şi faptul că prezenţa crescută a mutaţiilor 35delG este în strînsă legătură cu nivelul crescut de consangvinitate în cadrul populaţiilor respective, lucru ce nu se aplică în cazul populaţiilor europeane, respectiv românească.
În cadrul studiilor efectuate până în prezent, frecvenţa purtătorilor de mutaţie 35delG variază de la 1,1% în Iordania la 1,66% în Siria şi la 3,2 în Italia, cea mai mare frecvenţă cunoscută până în acest moment(1).
Rezultatele studiului de faţă sugerează faptul că prevalenţa crescută a mutaţiei 35delG în populaţia românească ar reprezenta premizele pentru instituirea diagnosticului şi sfatului genetic la un nivel extins, dar şi importanţa continuării investigaţiilor pe această temă. Tehnica utilizată în detecţia mutaţiilor este în continuare destul de laborioasă şi scumpă, iar uneori predispusă la erori de secvenţiere(10).
W24X este altă allelă relativ frecvent întâlnită în etiologia hipoacuziei neurosenzoriale non-sindromice, în special în populaţiile din nordul şi sudul Indiei(1,20,30). Această mutaţie a fost descoperită în studiul nostru cu o incidenţă de 10% homozigot şi 6,66% heterozigot digenic.
Diagnosticarea unui copil cu hipoacuzie include o întreagă baterie de teste pentru stabilirea etiologiei(9,26), dar în absenţa indiciilor pentru o suferinţă sindromică, testarea genei GJB2 este actualmente recomandată drept test iniţial(2,26) urmată de testarea pentru deleţia GJB6 în cazul unui test negativ(26,27). Aceste teste genetice au devenit o modalitatea foarte utilă de diagnosticare pentru specialistul O.R.L. în tentativa de a explica etiologia hipoacuziei depistate prin screening auditiv post-natal. Beneficiul principal al testării GJB2/GJB6 este faptul că, în anumite cazuri, poate explica apariţia hipoacuziei fără a mai fi necesare teste suplimetare(26,31), dar poate, de asemenea, aduce informaţii despre modul de transmitere al mutaţiei sau şansele de apariţie şi la posibilii urmaşi(26,31,36).
Cu toate că diagnosticul genetic este deosebit de costisitor şi nu este efectuat frecvent în ţările în curs de dezvoltare, investigarea mutaţiilor genei GJB2 devine esenţială pentru clarificarea problemei hipoacuziei congenitale. Acest diagnostic conduce la efectuarea sfatului genetic pentru membrii familiei şi facilitează reabilitarea precoce a copilului.
Concluzii
Studiul de faţă confirmă prezenţa într-un procent relativ mare a mutaţiei 35delG a genei GJB2 în cazurile de hipoacuzie neuro-senzorială congenitală non-sindromică profundă bilaterală, fapt ce este în concordanţă cu cifrele raportate în literatură. La doi pacienţi a fost descoperită o dublă mutaţie 35delG/W24X. Aceste rezultate subliniază importanţa diagnosticului genetic în clarificarea etiologiei şi în implementarea cât mai precoce a unui program de recuperare auditiv-lingvistică a pacientului. De asemenea, prin frecvenţa crescută a purtătorilor în populaţiile europene, se prefigurează necesitatea pentru screening-ul mutaţiei 35delG lucru ce ar facilita identificarea precoce a acestor purtători şi consilierea familială necesară în aceste cazuri.
Acknowledgments: Această lucrare a beneficiat de suport financiar prin proiectul „CERO - Cercetător roman de carieră”, POSDRU/159/1.5/S/135760 cofinanţat de Fondul Social European prin Programul Operaţional Sectorial Dezvoltarea Resurselor Umane 2007-2013./This work received financial support through the project entitled “CERO- Career profile: Romanian Researcher”, grant number POSDRU/159/1.5/S/135760 cofinanced by the European Social Fund for Sectoral Operational Programme Human Resources Development 2007-2013”.
Bibliografie
-
Al-Achkar W., Moassass F., Al-Halabi B., Al-Ablog A. Mutations of the Connexin 26 gene in families with non-syndromic hearing loss. Molecular Medicine REPORTS, 2011, 4: 331-35.
-
American College of Medical Genetics. Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genet Med 2002;4:162–171. [PubMed: 12180152].
-
Batissoco AC, Abreu-Silva RS, Braga MC, Lezirovitz K, Della-Rosa V, Alfredo T Jr, et al. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: implications for diagnosis and genetic counseling. Ear Hear. 2009;30(1):1-7.
-
Bochkov N. Clinical genetics: Manual, Moscow 2002.
-
Bork J., Peters L., Riazuddin S. Genetic and metabolic hearing disorders. American Journal of Human Genetic, 2001, 68:1; 26-37.
-
Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, Avan P, Cohen-Salmon M. Hearing is normal without Connexin 30. The Journal of Neuroscience. 2013, 33(2): 430-34.
-
Cordeiro-Silva MdF, Barbosa A, Santiago M, Provetti M, Rabbi- Bortolini E. Prevalence of 35delG/GJB2 and del (GJB6-D13S1830) mutations in patients with non-syndromic deafness from a population of Espírito Santo – Brazil. Braz J Otorhinolaryngol. 2010;76(4):428-32.
-
Cryns K, Orzan E, Murgia A, et al: Agenotype-phenotype correlation for GJB2 (connexin 26) deafness. J Med Genet 41: 147-154, 2004.
-
Duncan RD, Prucka S, Wiatrak BJ, Smith RJH, Robin NH. Pediatric otolaryngologists’ use of genetic testing. Arch Otolaryngol Head Neck Surg 2007;133:231–236. [PubMed: 17372079]
-
Frei K., Szuhai K., Lucas T., Weipoltshammer K., Schofer C., Ramsebner R., Baumgartner WD., Raap AK et.al. Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. European Journal of Human Genetics 2002, 10, 427 – 32.
-
Fuse Z., Doi K., Hasegawa T., Sugii A., Hibino H., Kubo T. Three novel connexin26 gene mutations in autosomal recessive nonsyndromic deafness, NeuroReport, 1999; 10(9), 1853–57.
-
Gasparini P, Rabionet R, Barbujani G et al.: High carrier frequency of the 35delG deafness mutation in European populations. Eur J Hum Genet 2000; 8: 19 – 23.
-
Gorlin, RJ.; Toriello, HV.; Cohen, MM, Jr. Hereditary Hearing Loss and Its Syndromes. New York: Oxford University Press; 1995.
-
Grifa A. , Wagner C.A., D’Ambrosio L. et al., Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus, Nature Genetics, 1999; 23(1), 16–18.
-
Hamid M, Karimipoor M, Hashemzadeh Chaleshtori M, Taghi Akbari M. A novel 355–357delGAG mutation and frequency of connexin-26 (GJB2) mutations in Iranian patients. Journal of Genetics, 2009, 88.3, 359-62
-
Hashemzadeh Chaleshtori M, Hoghooghi Rad L, Dolati M, et al: Frequencies of mutations in the connexin 26 gene (GJB2) in two populations of Iran (Tehran and Tabriz). Iranian J Publ Health 34: 1-7, 2005.
-
James, C., Hadley, D., Holtzman, A., Winkelstein, A.. How does the mode of inheritance of a genetic condition influence families? A study of guilt, blame, stigma, and understanding of inheritance and reproductive risks in families with X-linked and autosomal recessive diseases. Genetics in Medicine, 2006, 8, 234–42.
-
Janecke AR, Hirst-Stadlmann A, Günther B, Utermann B, Müller T, Löffler J, Utermann G and Nekahm-Heis D: Progressive hearing loss, and recurrent sudden sensorineural hearing loss associated with GJB2 mutations – phenotypic spectrum and frequencies of GJB2 mutations in Austria. Hum Genet 111: 145-153, 2002.
-
Koroleva I., Grigoreva I., Petriga E. Modern methods of differential diagnoses of hearing disorders. Materials of scientific-practical conference, 1999 Suzdal.
-
Maheswari M, Vijaya R, Ghosh M, Shastri S, Kabra M and Menon PSN: Screening of families with autosomal recessive nonsyndromic hearing impairment (ARNSHI) for mutations in GJB2 gene. Indian Scenario. Am J Med Genet 120: 180-184, 2003.
-
Markova T., Shagina I., Megrelishvilli el al. DNK-diagnostics congenital and early childhood hearing loss and deafness, Bulletin of otorhinolaryngology, 2002; 6, 12-15.
-
Mustaapha BM, Salem N, Delague V, et al.: Autosomal recessive non-syndromic hearing loss in the Lebanese population: prevalence of the 30delG mutation and report of two novel mutations in the connexin 26 (GJB2) gene. J Med Genet 38: E36, 2001.
-
Mustafa MW: Prevalence of the connexin 26 mutation 35delG in non-syndromic hearing loss in Egypt. Internet J Otorhinolar 3: http://www.ispub.com/ostia/index.php?xmlFilePath=journals/ijorl/vol3n1/connexin.xml, 2004.
-
Nance WE. The genetics of deafness. Ment Retard Dev Disabil Res Rev 2003;9:109–119. [PubMed:12784229]
-
Nogueira C, Coutinho M, Pereira C, Tessa A, Santorelli FM, Vilarinho L. Molecular Investigation of Pediatric Portuguese Patients with Sensorineural Hearing Loss. SAGE-Hindawi Access to Research Genetics Research International Volume 2011, Article ID 587602, 5 pagesdoi:10.4061/2011/587602.
-
Palmer C.G.S., Lueddeke J.T., Zhou J. Factors influencing parental decision about genetics evaluation for their deaf or hard-of-hearing child, Genet Med. 2009 April; 11(4): 248. doi:10.1097/GIM.0b013e318195aad9.
-
Pandya A., Arnos KS, Xia XJ et al. Frequency and distribution of GJB2 (connexin 26) and GJBG (connexin 30) mutations in a large North American repository of deaf probands. Genetics in Medicine, 2003, 5(4), 295–303.
-
Pfeilsticker LN, Stole G, Sartorato EL, Delfino D, Guerra ATM. A investigação genética na surdez hereditária não-sindrômica. Rev Bras Otorrinolaringol. 2004;70(2):181-6.
-
Piatto VB, Bertollo EM, Sartorato EL, Maniglia JV. Prevalence of the GJB2 mutations and the del(GJB6-D13S1830) mutation in Brazilian patients with deafness. Hear Res. 2004;196:87-93.
-
Ramshankar M, Girirajan S, Dagan O, Ravi Shankar HM, Jalvi R, Rangasayee R, Avraham KB and Anand A: Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J Med Genet 40: E68, 2003.
-
Schimmenti LA, Martinez A, Fox M, Crandall B, Shapiro N, Telatar M, et al. Genetic testing as part of the Early Hearing Detection and Intervention (EHDI) process. Genet Med 2004;6:521–525. [PubMed: 15545749]
-
Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, and Kanaan M: Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet 110: 284-289, 2002.
-
Smith, R., Hildebrand, M., Van Camp, G. (2010). Deafness and hereditary deafness overview. http://www.ncbi.nlm.nih.gov/pubmed/20301607.
-
Steinberg, A. G., Kaimal, G., Bain, L., Krantz, I., Li, Y. (2007). Parental narratives on genetic testing for children with deafness: a qualitative inquiry. American Journal of Medical Genetics, 143(A),1533–1545.
-
Wilcox SA, Osborn AH and Dahl HH: Simple PCRtest to detect the common 35delG mutation in the connexin 26 gene. Mol Diagn 5: 75-78, 2000.
-
Withrow KA, Burton S, Arnos KA, Kalfoglou A, Pandya A. Consumer motivations for pursuing genetic testing and their preferences for the provision of genetic services for hearing loss. J Genet Counsel 2008;17:252–260.
-
Worldwide survey of the delta F508 mutation: Report from the cystic fibrosis genetic analysis consortium. Am J Hum Genet 1990; 47: 354 – 359.
-
Zelante L., Gasparini P., Estivill X. et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Human Molecular Genetics. 1997; 6(9); 1605–09.

Displazie fibroasă de sfenoid drept – caz clinic
Bogdan Mocanu, Silviu Oprescu, Alina Ciocâlteu, Emilia Diaconu
Autorii prezintă cazul unei paciente de 32 de ani cu sindrom cefalalgic sever, neresponsiv la tratamentul medicamentos specific, scăderea acuităţii vizuale la ochiul drept şi diplopie, simptomatologie oculară apărută epi...
Rezumate Conferinţa ORL.ro
Rezumate Conferinţa ORL.ro...
Displazie fibroasă fronto-orbitală dreaptă
Bogdan Mocanu, Silviu Oprescu, Irina Boagiu, Denisse Creţu, A. Coman, Daniel Mirea, Vlad Andrei Budu
...
Study models for salivary gland regeneration
Mihnea Ioan Nicolescu
Glandele salivare prezintă o patologie degenerativă complexă, cu răsunet important asupra calităţii vieţii. Cauzele reducerii funcţionale sunt multiple, de la procese tumorale la boli autoimune. ...
From hirsutism and menstrual issues to congenital adrenal hyperplasia: is genetic testing a stepping stone for 3-beta-hydroxysteroid dehydrogenase type 2 deficiency?
Florica Şandru, Ana-Maria Gheorghe, Eugenia Petrova, Răzvan Petca, Mihai-Cristian Dumitraşcu, Claudiu-Eduard Nistor, Mara Carsote
Hiperplazia adrenală congenitală (CAH) este o provocare, iar în cazurile cu debut tardiv şi fenotip uşor care nu au fost detectate...