Cardiopatie dilatativă la un adolescent – prezentare de caz
Dilated cardiomyopathy in an adolescent – a diagnostic challenge
Abstract
Background. Heart failure is a rare but serious diagnosis in pediatric patients. While in newborns and infants congenital heart disease is commonly the cause, in older children and in adolescents cardiomyopathies (primary or secondary) take the lead. The diagnosis may be difficult to establish, as the symptoms are nonspecific (especially in young ages). Case presentation. A previously healthy 16-year-old girl had been experiencing heart failure symptoms for the past 10 months, with gradual worsening of symptom severity. Upon evaluation, a diagnosis of class III Ross/NYHA heart failure due to dilated cardiomyopathy with severe left ventricular systolic dysfunction was established. Standard heart failure medication was initiated, alleviating congestion and partially controlling symptoms. There was no family or personal history of heart disease. The patient reported excellent exercise tolerance previously. No exposure to potentially toxic agents has been identified. The initial tests could not establish a definite cause, although a possible respiratory tract infection at the onset of symptoms and certain cardiac magnetic resonance imaging features could suggest previous eosinophilic myocarditis. Current literature to guide the diagnosis process and treatment options are discussed. Conclusions. Dilated cardiomyopathy is rare but severe in children. Even the typical symptoms may be downplayed or ignored for long periods, delaying the diagnosis; a high index of suspicion is therefore required when other more common disorders seem unlikely or have been ruled out. The possible underlying causes vary with age and differ from the adult population. An etiological diagnosis is frequently difficult to achieve, but must nonetheless be sought out, as it may allow for targeted therapy and improve prognosis. However, long-term outcome is usually grim.Keywords
pediatricchildadolescentheart failuredilated cardiomyopathyRezumat
Introducere. Insuficienţa cardiacă este un diagnostic rar, însă sever în populaţia pediatrică. Dacă la nou-născut şi sugar principala cauză o reprezintă bolile cardiace congenitale, la copilul mare şi adolescent se regăsesc în prim-plan cardiomiopatiile primare sau secundare. Diagnosticul poate fi dificil de stabilit, simptomele fiind nespecifice (în special la vârstele mici). Prezentarea cazului. O pacientă în vârstă de 16 ani, fără antecedente semnficative, prezintă de aproximativ 10 luni simptomatologie compatibilă cu diagnosticul de insuficienţă cardiacă, agravată progresiv. În urma evaluării se pune diagnosticul de insuficienţă cardiacă clasa III Ross/NYHA prin cardiomiopatie dilatativă, cu disfuncţie sistolică severă de ventricul stâng. Se iniţiază terapie medicamentoasă specifică, obţinându-se ameliorarea fenomenelor de congestie şi controlul parţial al simptomelor. Adolescenta nu prezenta istoric familial sau personal de boală cardiacă şi afirmă o toleranţă excelentă la efort anterior epsiodului descris. Nu s-a identificat expunere la agenţi cu potenţial cardiotoxic. În baza investigaţiilor iniţiale nu s-a putut stabili un diagnostic etiologic cert, însă un posibil episod de infecţie respiratorie la debutul simptomatologiei, precum şi aspectul imagistic la rezonanţa magnetică cardiacă ar putea sugera un episod de miocardită eozinofilică în antecedente. Am analizat literatura relevantă recentă şi opţiunile de tratament. Concluzii. Cardiomiopatia dilatativă este rară, dar severă la copil. Chiar şi simptomele tipice pot fi subestimate sau ignorate mult timp, întârziind astfel diagnosticul; se impune deci un nivel ridicat de suspiciune în momentul în care alte afecţiuni mai frecvente sunt improbabile sau au fost infirmate. Posibilele cauze variază în funcţie de vârstă şi sunt diferite de cele ale adultului. Diagnosticul etiologic este în mod frecvent dificil de stabilit, însă trebuie căutat deoarece poate permite terapia ţintită. Prognosticul pe termen lung rămâne de regulă rezervat.Cuvinte Cheie
pediatriccopiladolescentinsuficienţă cardiacăcardiomiopatie dilatativăBackground
Heart failure is a rare but serious diagnosis in pediatric patients. Both symptoms and more frequent causes will vary according to age group. While in newborns and infants structural heart disease is commonly the cause, in older children and in adolescents cardiomyopathies (primary or secondary) take the lead. The diagnosis may be difficult to establish, as the symptoms are nonspecific (especially at young ages), but even in cases with typical symptoms the diagnosis is sometimes delayed due to its rarity.
Case presentation
A previously healthy 16-year-old girl had been experiencing fatigue and exertional dyspnea for the past 10 months. Her exercise tolerance had decreased slowly but consistently over this period. For the past month, she also reported nocturnal and supine cough, decreased appetite and involuntary weight loss of around 1.5 kg. Two weeks previously she started experiencing nausea, abdominal discomfort and postprandial vomiting, which prompted a doctor appointment.
The patient had no significant medical history and was not taking any medication. She had been a professional athlete for some years; she had interrupted training several years previously, but not for medical reasons. Both her parents and her younger sibling were healthy.
Upon examination, her height and weight were normal for her age. She had regular, albeit tachycardic heart sounds (115 beats per minute) with occasional extra beats and no murmurs, her blood pressure was 95/60 mmHg, there was no peripheral edema, the liver edge was at approximately 2 cm below the costal margin, and she was experiencing mild discomfort on palpation. Hepatojugular reflux was noted. Her chest was clear and her peripheral oxygen saturation was normal on room air.
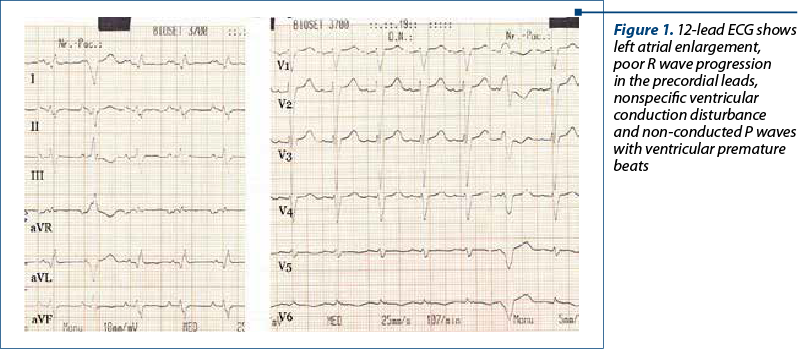
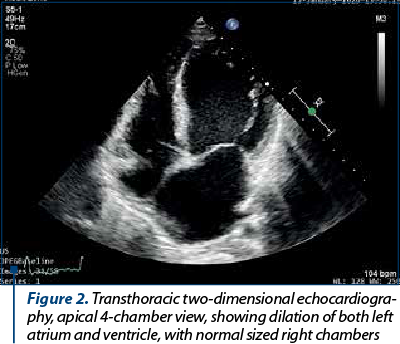
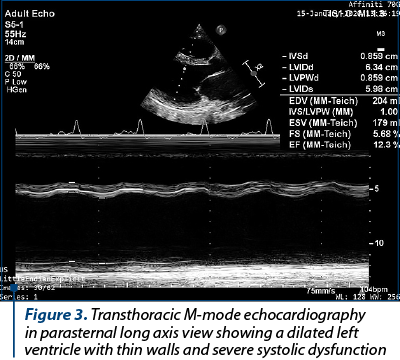
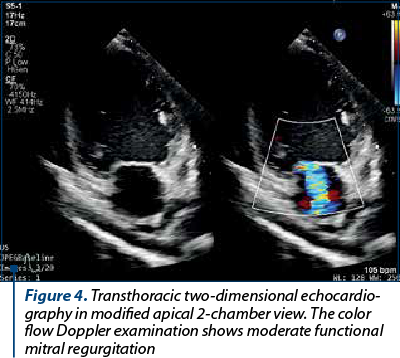
Her ECG showed left atrial enlargement, poor R wave progression in the precordial leads, nonspecific ventricular conduction disturbance and ventricular premature beats (VPBs) – Figure 1. A chest X-ray had been performed previously and reported an enlarged heart silhouette, but no pulmonary abnormalities. An echocardiography was performed and showed a dilated left ventricle (LV) with severely impaired systolic function (the ejection fraction [EF] was visually estimated at 15% and calculated at 25% using Simpson’s biplane method) – Figures 2 and 3. The LV walls were thin and diffusely hypokinetic; the wall motion abnormality seemed to be worst at the level of the lateral wall, which appeared akinetic. The left atrium was also dilated. There was moderate functional mitral regurgitation (Figure 4) on color flow Doppler examination. The right heart did not appear dilated and right ventricular (RV) longitudinal motion was preserved, with a tricuspid annulus plane systolic excursion (TAPSE) of 18 mm. The pulmonary artery systolic pressure was estimated at 55 mmHg. There was a small amount of pericardial effusion located posteriorly. The origin of the coronary artery system appeared normal.
Initial blood tests showed mild anemia, but no iron deficit (interpreted as possibly due to dilution secondary to systemic fluid overload), mild vitamin D deficiency, and a slightly increased international normalized ratio (INR). The N-terminal pro-BNP (NT-proBNP) was elevated at over 5000 pg/ml. Other tests, including C-reactive protein, electrolytes, venous blood gas test, renal, liver and thyroid function tests, were normal. The 24-hour ECG monitoring was performed and showed rare VPBs, mostly isolated, but also periods of trigeminy and bigeminy. No sustained arrhythmia or atrioventricular conduction abnormalities was noted.
Based on initial tests, a diagnosis of class III Ross/NYHA heart failure (HF) caused by a dilated cardiomyopathy (DCM) with severely impaired LV systolic function was established. The medical therapy was initiated, including diuretics (mineralocorticoid receptor [MR] antagonist and loop diuretic) and very low-dose beta-blocker (carvedilol 3.125 mg per day). Congestion was well controlled and digestive symptoms subsided. The constantly low (and occasionally symptomatic) systolic BP averaging 80-90 mmHg precluded the initiation of angiotensin-converting-enzyme inhibitors (ACE-Is). Vitamin D, carnitine and coenzyme Q10 oral supplements were also administered. The patient was subsequently referred for specific assessment for heart transplantation.
The initial evaluation eliminated structural cardiac disease, active infection, hematologic disorders, anemia or severe calcium, phosphorus or magnesium deficits, thyroid or renal dysfunction or muscular dystrophies as potential causes. A tachycardia-induced cardiomyopathy seemed unlikely, as no sustained arrhythmias were recorded. The family reported no exposure to potentially toxic agents. An episode of upper respiratory tract infection may have been present at the onset of symptoms.
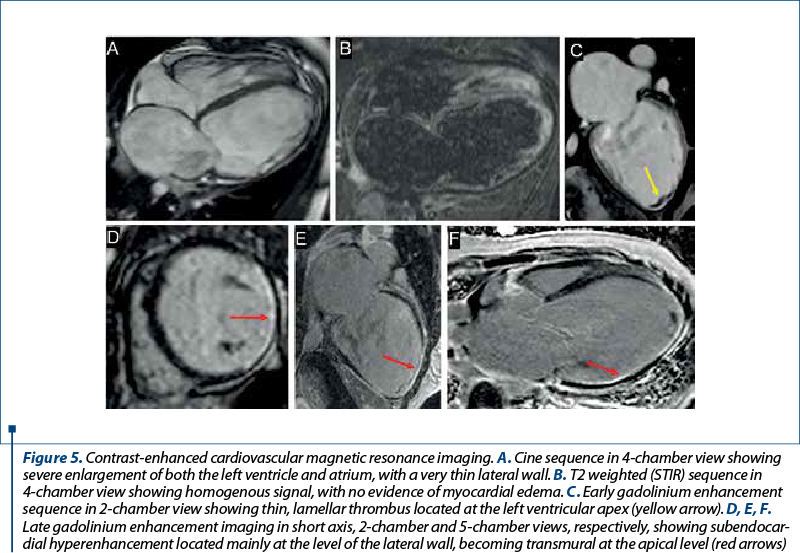
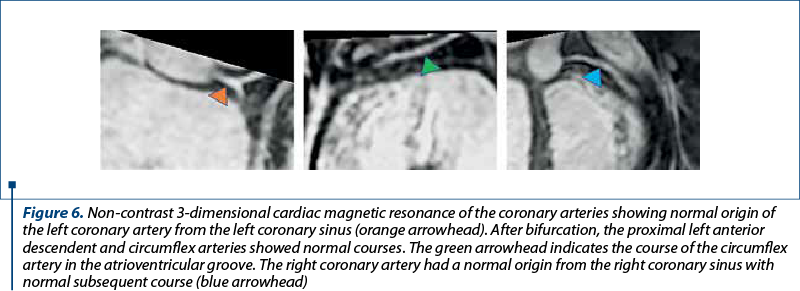
Cardiac magnetic resonance (CMR) imaging confirmed the echocardiographic findings: a severely dilated LV with severe systolic dysfunction (EF 12%) due to global hypokinesia and akinesia of the lateral wall, which was also thinner (Figure 5A). Although the RV was not dilated, its systolic function was severely impaired (RV EF 23%), mainly due to absence of transversal contraction and akinesia of the apical segments, with relatively preserved longitudinal contraction of the subtricuspidian segments. There was no evidence of myocardial edema and thus no clear criteria for active myocardial inflammation (Figure 5B). A small, lamellar thrombus was noted at the LV apex on early gadolinium enhancement imaging, a finding which prompted anticoagulation (Figure 5C). Late gadolinium enhancement (LGE) imaging showed extensive subendocardial fibrosis located mainly at the lateral wall level, but extending in a transmural fashion at the level of the apical segments of the septum, anterior and posterior walls (Figure 5D, E and F). No scar was seen at the basal level of the interventricular septum, as well as on the anterior and inferior walls. The origin and proximal course of the coronary arteries were confirmed to be normal (Figure 6). The subendocardial positioning of the fibrosis raised the suspicion of hypereosynophilic myocarditis; for the same reason, however, coronary heart disease could not be categorically excluded, although the scar area seemed to extend beyond the territory of the circumflex artery.
At this stage, no definite etiology could be established, although the possible episode of respiratory infection at the (or slightly before) the onset of symptoms and the pattern of fibrosis described by CMR imaging could suggest previous eosinophilic myocarditis.
Discussion
Heart failure symptoms may be elusive during childhood and vary with age. Children and adolescents in particular have a great capacity of compensating for cardiac dysfunction for a long time and even typical symptoms may be erroneously dismissed or attributed to other causes (such as detraining or lack of exercise, excessive heat and humidity during summer, mild respiratory infections etc.), especially if the worsening is gradual over a long period. In our case, the diagnosis was only established 10 months after the onset of symptoms. The etiology also varies with age, with structural heart disease (and congenital defects in particular) dominating the scene in newborns and infants. This changes in older children, with cardiomyopathies, either primary or secondary, becoming the most common cause(1).
Cardiomyopathies are rare during childhood, affecting around 1 in 100,000 children(2), and with an incidence of around 1 per 100,000 person-years for primary cardiomyopathies, according to various studies(3-6). However, the prognosis is severe, with an estimated nearly 40% of symptomatic children requiring heart transplantation or dying within two years from the diagnosis(4,7,8). DCM makes for around 50% of pediatric cardiomyopathy cases(6).
The search for an etiology may be extremely cumbersome when the cause is not readily apparent; however, if a cause can be established, it may greatly improve the prognosis in these patients, as it allows for targeted therapy. Most reviews and guidelines(6,9-13) recommend a similar approach, with an ECG, chest X-ray, routine blood tests and baseline B-type natriuretic peptide (BNP) or NT-proBNP and detailed echocardiography being the first step.
CMR is considered the gold standard for heart chamber quantification due to its high resolution and is routinely recommended in the workup of cardiomyopathies for its tissue characterization properties. In our case, the CMR exam confirmed the LV dilatation and severe dysfunction and additionally identified RV dysfunction. The drawbacks of echocardiographic RV function quantification are well known, as well as the high accuracy of CMR for this specific indication. The CMR also identified the LV apical thrombus, which had not been diagnosed using the non-contrast echocardiography examination. Furthermore, the LGE imaging identified subendocardial scarring of the lateral wall as being the cause of the thinner and akinetic lateral wall compared to the rest of the LV segments. In theory, any subendocardial scar raises the suspicion of ischemia, prompting a thorough investigation of the coronary anatomy. Congenital anomalies of the origins of the coronary arteries (such as the ALCAPA syndrome) are an important cause of LV dysfunction. However, as the 3D CMR showed, the origins and proximal course of the three coronaries were normal. Moreover, the scar seemed to extend beyond the circumflex artery territory, making ischemic heart disease less probable. Another explanation for the subendocardial scarring may be a previous hypereosinophilic myocarditis. While CMR imaging may raise the suspicion, such an etiology needs to be confirmed by endomyocardial biopsy, especially when the peripheral blood count does not show eosinophilia.
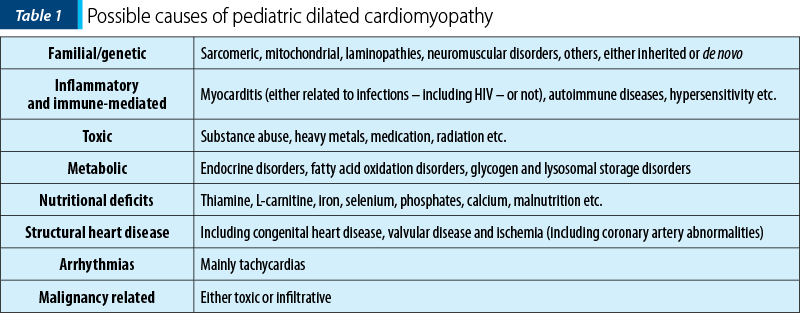
Depending on the initial results, additional blood tests, genetic testing and various other imaging modalities (such as stress tests, cardiac catheterization – mainly used as part of pre-transplant assessment, but also for hemodynamic or coronary anatomy assessment, myocardial biopsy etc.), as well as a multidisciplinary team approach may be necessary. In some cases, the identification of a cause is not possible and the DCM is labeled as idiopathic. The possible causes of pediatric DCM are listed in Table 1.
In the absence of an underlying cause, the chronic treatment aiming to control symptoms and improve prognosis consists of well-established drugs such as ACE-Is, beta-blockers and MR antagonists. In addition, in recent years, attempts have been made to align the pediatric medical treatment options to those available for adults. A recent study reports the use of ivabradine, safely reducing the resting heart rate of children with chronic HF and DCM(14). Recently, the U.S. Food and Drug Administration (FDA) has approved the use of an angiotensin receptor neprilysin inhibitor for pediatric use(15) (sacubitril/valsartan, a combination that showed excellent results in adults with symptomatic HF with a reduced EF(16), and is currently being tested in pediatric patients(17)).
Last but not least, the risk of sudden cardiac death, although smaller for DCM compared to other cardiomyopathies, is not to be ignored. Some risk factors have been identified and are summarized in recent literature(18). However, in the absence of dedicated pediatric guidelines, adult recommendations for preventive use of implantable defibrillators (such as the European and American guidelines(19,20)) are usually employed.
Conclusions
DCM is a rare but severe diagnosis in children. Symptoms, even when typical, may be downplayed or ignored for long periods, delaying the diagnosis; a high index of suspicion is therefore required when other more common disorders seem unlikely or have been ruled out.
The possible underlying causes vary with age and differ from the adult population. An etiological diagnosis is frequently difficult to achieve, but must nonetheless be sought out, as it may allow for targeted therapy and improve prognosis. However, the long-term outcome is usually grim.
Bibliografie
-
Price JF. Congestive heart failure in children. Pediatr Rev. 2019 Feb 1; 40(2):60–70.
-
Wilkinson JD, Westphal JA, Bansal N, Czachor JD, Razoky H, Lipshultz SE. Lessons learned from the Pediatric Cardiomyopathy Registry (PCMR) Study Group. In: Cardiology in the Young. Cambridge University Press. 2015; p. 140–53.
-
Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents: A nationwide study in Finland. Am J Epidemiol [Internet]. 1997 Sep 1 [cited 2020 Apr 25]; 146(5):385–93. Available from: https://academic.oup.com/aje/article-lookup/doi/10.1093/oxfordjournals.aje.a009291
-
Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003 Apr 24; 348(17):1647–55.
-
Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003 Apr 24; 348(17):1639–46.
-
Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation. 2019 Jul 2; 140(1):E9–68.
-
Alexander PMA, Daubeney PEF, Nugent AW, Lee KJ, Turner C, Colan SD, et al. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: Results from a national population-based study of childhood cardiomyopathy. Circulation [Internet]. 2013 Oct 29 [cited 2020 Apr 25]; 128(18):2039–46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24036608
-
Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, et al. Natural History of Dilated Cardiomyopathy in Children. J Am Heart Assoc [Internet]. 2016 Jul 6 [cited 2020 Apr 25]; 5(7). Available from: https://www.ahajournals.org/doi/10.1161/JAHA.116.003450
-
Kantor PF, Lougheed J, Dancea A, McGillion M, Barbosa N, Chan C, et al. Presentation, diagnosis, and medical management of heart failure in children: Canadian cardiovascular society guidelines. Can J Cardiol. 2013 Dec 1; 29(12):1535–52.
-
Kirk R, Dipchand AI, Rosenthal DN, Addonizio L, Burch M, Chrisant M, et al. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. J Hear Lung Transplant. 2014 Sep 1; 33(9):888–909.
-
Jayaprasad N. Heart failure in children. Heart Views. 2016; 17(3):92–9.
-
Masarone D, Valente F, Rubino M, Vastarella R, Gravino R, Rea A, et al. Pediatric Heart Failure: A Practical Guide to Diagnosis and Management. Pediatr Neonatol. 2017 Aug 1; 58(4):303–12.
-
Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Pediatric cardiomyopathies. Circ Res. 2017 Sep 1; 121(7):855–73.
-
Bonnet D, Berger F, Jokinen E, Kantor PF, Daubeney PEF. Ivabradine in Children With Dilated Cardiomyopathy and Symptomatic Chronic Heart Failure. J Am Coll Cardiol. 2017 Sep 5; 70(10):1262–72.
-
Novartis. Novartis Entresto receives FDA approval for pediatric heart failure, helping to address critical unmet need for treatment options. Novartis US [Internet]. 2019 [cited 2020 Apr 25]. Available from: https://www.novartis.us/news/media-releases/novartis-entresto-receives-fda-approval-pediatric-heart-failure-helping-address
-
McMurray JJV, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014 Sep 11; 371(11):993–1004.
-
Shaddy R, Canter C, Halnon N, Kochilas L, Rossano J, Bonnet D, et al. Design for the sacubitril/valsartan (LCZ696) compared with enalapril study of pediatric patients with heart failure due to systemic left ventricle systolic dysfunction (PANORAMA-HF study). Am Heart J. 2017 Nov 1; 193:23–34.
-
Rella V, Parati G, Crotti L. Sudden Cardiac Death in Children Affected by Cardiomyopathies: An Update on Risk Factors and Indications at Transvenous or Subcutaneous Implantable Defibrillators. Front Pediatr. 2020 Apr 3; 8:139.
-
Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac deathThe Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European. Eur Heart J [Internet]. 2015 Nov 1; 36(41):2793–867. Available from: http://dx.doi.org/10.1093/eurheartj/ehv316
-
Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation. 2018 Sep 25; 138(13):e272–391.

Capcane de diagnostic în patologia pulmonară malformativă la copil
Alina Murgu, Bianca Bendoiu, Gianina Darie, Ionela Dobre, Flavia Hoşleag, Ioana Cernescu, Doina Nedelcu, Bogdan A. Stana
Bronchogenic cyst belongs to congenital non-vascular lung malformations....
Sindrom Lyell - consideraţii pe baza unui caz clinic
Alice Azoicăi, Cristiana Andronic, Bogdan A. Stana
Lyell syndrome – or toxic epidermal necrolysis – is a severe mucocutaneous reaction caused by the administration of a drug, and is characterized by a rash of the epidermis that fuses rapidly and turns into a large dark erythema, with intense necrosis and scaling of the epidermis in the cloth, similar to skin ...
Trisomia 18 – cauză rară de malformaţie cardiacă complexă
Mirela Silvia Iancu, Daniela Popeia, Varvara Toma, Ana Maria Daviţoiu, Victoria Hurduc, Doina Anca Pleşca
Trisomy 18 (Edwards syndrome), a rare genetic condition, recognized since 1960, is the second most common autosomal trisomy, after trisomy 21....
Capcane de diagnostic în patologia pulmonară malformativă la copil
Alina Murgu, Bianca Bendoiu, Gianina Darie, Ionela Dobre, Flavia Hoşleag, Ioana Cernescu, Doina Nedelcu, Bogdan A. Stana
Bronchogenic cyst belongs to congenital non-vascular lung malformations....
Sindrom Lyell - consideraţii pe baza unui caz clinic
Alice Azoicăi, Cristiana Andronic, Bogdan A. Stana
Lyell syndrome – or toxic epidermal necrolysis – is a severe mucocutaneous reaction caused by the administration of a drug, and is characterized by a rash of the epidermis that fuses rapidly and turns into a large dark erythema, with intense necrosis and scaling of the epidermis in the cloth, similar to skin ...