Emergencies in rheumatic diseases in children (II): antiphospholipid syndrome
Urgenţe în reumatologia pediatrică (II): sindromul antifosfolipidic
Abstract
Sometimes, child’s rheumatic diseases are generating emergency type complications which, if not diagnosed in time, are life-threatening. Antiphospholipid syndrome in its severe form can induce coagulation disorders and acute organ failure, depending on location and extent. Systemic lupus erythematosus is frequently associated with this pathology and must be carefully investigated and monitored in the evolution of the patient, in order to benefit from early specific anticoagulant treatment, associated with chronic background immunosuppressive treatment.Keywords
childantiphospholipid syndromeemergencysystemic lupusRezumat
Uneori, bolile reumatismale ale copilului conduc la complicaţii de tip urgenţă care, nediagnosticate la timp, pot pune în pericol viaţa acestuia. Sindromul antifosfolipidic în forma sa severă poate induce tulburări de coagulare şi insuficienţă acută de organ, în funcţie de localizare şi de gradul de extindere. Lupusul eritematos sistemic asociază frecvent această patologie, care trebuie investigată şi monitorizată atent în evoluţia bolnavului, pentru a putea beneficia de tratament precoce specific anticoagulant, asociat la tratamentul imunosupresiv cronic de fond.Cuvinte Cheie
copilsindrom antifosfolipidicurgenţălupus sistemicIntroduction
One of the causes that induce thromboembolic coagulation disorders in children’s rheumatic diseases is antiphospholipid syndrome (APS). In its severe form, it constitutes an emergency pathology that puts the patient’s life at risk. The clinical manifestations are frequently similar to those of sepsis, rather than rheumatic disease, especially in the early period, so that the risk of misdiagnosis and late introduction of the correct life-saving treatment is major and with prognostic impact.
Clinical case
The patient P.S., male, 14 years old, from urban area, presented in the emergency department with pain and sensation of chest compression with cervical irradiation, severe dyspnea, orthopnea and fever (39°C), with onset approximately 3-4 days prior to presentation. At home, he received paracetamol treatment alternating with non-steroidal anti-inflammatory drugs (NSAIDs), without the improvement of symptoms.
Anamnestically, we mention the diagnosis of immune thrombocytopenic purpura (four years before), frequent acute respiratory tract infections, “convulsive” cough and acute pneumopathy treated with antibiotics at home one month before. Family history highlighted maternal grandmother diagnosed with autoimmune hemolytic anemia.
Clinical examination: severe general condition, febrile (39°C), cutaneous pallor, mixed dyspnea and orthopnea, intercostal draught, abolition of vesicular murmur at bilateral lung bases and dullness at this level, precordial pain, turgid jugulars, BP 105/63 mmHg, HF 105/min, SatO2 90-92% in atmospheric air, RF 25/min, distended abdomen, hepatomegaly (4 cm below the rim), splenomegaly (3 cm below the rim), physiological bowel movements, urine with normal aspect, normal neurological examination.
Biological investigations: Hb 8.1 g/dl, WC 4120/mmc, Tr 118,000/mmc, RC 3,8 x1012/l, R 46%, PMN 40%, L 40%, M 18.9%, E 0.7%, MCV 72.6fl, MCH 23 pg, MCHC 31.4 g/dl; peripheral blood smear – hypochromia, anisocytosis; significant inflammatory syndrome – fibrinogen 722 mg/dl, CRP 213.12 mg/dl, ESR 112mm/h, TGP 4, TGO 14 U/L, AP 186 U/l, TB 0.84 mg/dl, urea 31 mg/dl, creatinine 0.71 mg/dl, uric acid 3.27 mg/dl, glycemia 98 mg/dl, CPK 21 U/l, LDH 477U/l, total cholesterol 100 mg/dl, TG 110 mg/dl, amylase 28 U/L, TP 65.96 g/l, albumin 35.4 g/l, alpha-1 4.6 g/l, alpha-2 8.2g/l, ß1 5 g/l, ß2 2.8 g/l, у 10 g/l; urinary sediment analysis – frequent urates; ketone bodies ++, AR 21.43, Na 134.8, K 4.09 mmol/l; Addis test – C 0, H 2222, L 4444/min; urine 24 hours – Cl creatinine, density – normal values; proteinuria – absent, coproculture, uroculture and blood culture along with procalcitonin – negative.
Chest X-ray revealed interstitial intercleidohilar and right hilum infiltrate, bilateral costomarginal pleural reaction 4 cm thick on the right; cord with increased cross-sectional diameter on account of left lower arch; normal vascular pedicle; left middle arch obliteration.
Abdominal ultrasonography revealed homogeneous liver with normal reflectivity, right hepatic lobe with slightly increased size (139 mm), left hepatic lobe 69 mm, portal vein 10 mm in diameter, cholecyst, normal pancreas, spleen with increased bipolar diameter (160 mm), large amount of perihepatic fluid between loops, right and left iliac fossa, parietocolic and Douglas space.
EKG revealed sinus rhythm 100-109/min, AQRS+, PQ 0.12 s, and negative T waves in V1-V5. Doppler echocardiography revealed fluid in pericardial cavity in medium quantity, contraction asynchrony at interventricular septum, along with dilated jugular and hepatic veins (Figure 1 a, b).
Biological investigations: HBsAg, HCV-Ag, HIV-Ag – absent, Quantiferon TB Gold 0.01 UI/ml (<0.35) – negative test, IDR 5 IU PPD – negative, alpha fetoprotein 0.31 UI/ml (<5), RF<8 UI/ml.
Medulogram – normoplastic polymorphous marrow, normochromic erythropoiesis; efficient granulopoiesis maturing to end elements with mild eosinophilia; platelet megakaryopoiesis; platelets present on smear in clumps; relatively numerous plasma cells and monocytes.
At pericardial puncture, 5 ml of serum hematic fluid were extracted. Direct examination detected rare destroyed PMN, rare lymphocytes, frequent red blood cells, culture (including for Koch bacillus) – negative. Cytology: PMN 86%, lymphocytes 8%, M 2%, mesothelial cells 2%. Biochemistry: glucose 55 g/l, low total protein 48 g/l, LDH 2590 U/l, hypoalbuminemia (27.9), alpha-1 globulin 3, alpha-2 globulin 4.9, ß1 globulin 3.6, ß2 globulin 2, gamma globulin 7 g/l.
Pleural puncture was also performed, with the extraction of 30 ml of serocitrin fluid. Direct microscopic examination revealed rare intact PMN, frequent mesothelial cells, bacterioscopic negative; culture, including Koch bacillus – negative. Biochemically, the fluid was exudate-like, protein 37.04 g/l, glucose 103 mg/dl, LDH 372 U/l, ADA (adenosindeaminase) 23 U/l (<50). Pleural fluid was analyzed immunophenotypically, where the following cell populations were identified, without the presence of atypical lymphocytes: 12% lymphocytes (10% T lymphocytes, CD4+/CD8+ ratio = 0.5, 0.5% NK lymphocytes, 1.5% B lymphocytes without polyclonal atypia, kappa+/lambda+ ratio = 1.1), 13% monocytes, 75% granulocytes.
RHEUMA-ANA 9 total autoimmune disease profile detected dsDNA (x7 VN) 273.81 UI/ml (0-40), ssDNA (x1.4VN) 138 (<99), Ac anti Sm123↑ (<89), RNP/Sm 61 (<83), SSA (Ro) 46 (<91), SSB (La) 58 (<73), Scl 70 11 (<32), centromere 29 (< 100), chromatin 39 (<99); AAN positive. Profile for APS-Ac anticardiolipin: IgG 73 (x3.2 NV) (<23 GPL/ml), ß2GP1 (IgA, G, M) 49 (<20 RE/ml), D dimers 5000 (x25 NV), subsequently 573 ng/ml (0-250), C3 70.3 mg/dl (85-160), C4 31 mg/dl (10-40), protein S (activity) 77% (54-110%), protein C (activity) 81% (70-140%), factor V Leiden mutation not identified.
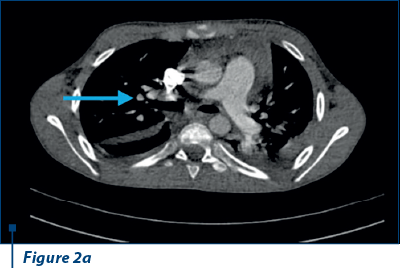
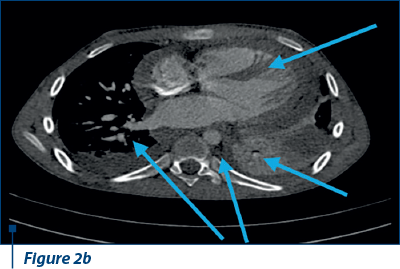
Thoracoabdominal CT angiography revealed microthrombosis in LMD segmental arteries; LID segment IV, pleural, pericardial fluid and left basal condensation focus (Figure 2 a, b).
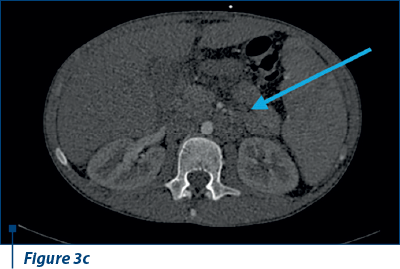
Also, ascites in large amounts, right bronchopulmonary adenopathy and adenopathy in the splenic hilum were evident (Figure 3 a, b, c).
Anamnestic data associated with clinical examination, laboratory investigations and imaging established the following diagnoses:
1. Systemic lupus erythematosus without renal involvement.
2. Secondary antiphospholipid syndrome.
3. Pulmonary thromboembolism.
3. Polyserositis (pericarditis; pleurisy; ascites).
4. Cardiac tamponade.
5. Left basilar pneumonia.
Double broad-spectrum antibiotic therapy (meropenem and teicoplanin), intravenous pulses of methylprednisolone, followed by chronic therapy with prednisone and azathioprine associated with anticoagulants (heparin initially, then Fraxiparine®), symptomatic treatment (hepatoprotective agents, proton pump inhibitors, short-course diuretics, antithermics), hygienic and dietary regime were instituted.
Discussion
Pediatric antiphospholipid syndrome is most commonly secondary to rheumatic pathological conditions, with an increased incidence in systemic lupus erythematosus (SLE; 50-83%), rheumatoid or psoriatic arthritis (33%) Sjögren’s syndrome (42%), vasculitis (20%), mixed connective tissue disease (22%), dermatomyositis, but also in other autoimmune pathologies such as thrombocytopenic purpura (30%), hemolytic anemia, infections (bacterial infections, HIV, hepatitis C etc.) or drug-induced (e.g., neuroleptics, antiarrhythmics, some antibiotics, interferon alpha etc.)(1). The pathogenesis of the disease is centered on the reaction of anti-complex binding protein antibodies (Beta 2 GP1, prothrombin), phospholipids, as well as cellular cofactors (annexin A2, apolipoprotein E receptor 2, Toll-like cell receptors 2 and 4) which induce endothelial, monocyte and neutrophil activation, as well as the complement system, with a role in the initiation and maintenance of inflammation and thrombosis(2,3). Currently, genetic involvement is also demonstrated in non-familial APS (HLA DQ/DR haplotypes, STAT 4 gene mutations)(4). Consequently, the clinical expression accompanying organ dysfunction is more frequent than in arterial thrombosis, depending on the location of the venous thrombosis, with predilection in the limbs, vena cavae, pulmonary, hepatic (Budd-Chiari syndrome), but also cerebral (cavernous sinus) or retinal vessels. A particularity of the child pathology is the incidence of primary (no specified cause) forms of LFS (20%), which subsequently demonstrate in some cases a progression to secondary APS in a specified etiological context (more common in SLE)(5,6).
The Sapporo diagnostic criteria (revised in 2006, Sydney, which are not specific for pediatric APS) associate with clinical data the positivity of antiphospholipid antibodies (anticardiolipin, anti-beta 2 glycoprotein 1) which have a certain frequency during the evolution of the disease, some of which are sometimes not objectified in the early stages (beta 2 glycoprotein 1 antibodies)(7,8). Although APS may have a mild or moderate form, the severe (catastrophic) aspect induces a mortality rate of over 50% through thrombotic microangiopathy, leading to multiple organ failure (persistent inflammatory syndrome, thrombocytopenia, hemolytic anemia, disseminated coagulopathy).
The therapeutic management includes chronic therapy with anticoagulants and/or platelet antiaggregants and systemic corticosteroids associated or not with other immunosuppressants (cyclophosphamide), the combination depending on the clinical-biological evolution and the response level to treatment. With encouraging results, there are approached in difficult situations or therapeutic nonresponse the following: plasmapheresis, administration of immunoglobulins (i.v.) or in patients with recurrent thrombosis, anti-CD20 (rituximab) or anti-C5 (eculizumab) therapy(9). The particularity of the case presented is the evolution of an immune thrombocytopenia years later, towards severe SLE with acute onset, manifested by polyserositis and acute pneumopathy, possibly caused by the association with severe pulmonary thrombosis-inducing APS(10,11).
In conclusion, SAPL, most commonly associated with pediatric rheumatic diseases, can induce an onset, but also a severe evolution, can occur at any stage of the underlying disease, causing loss of disease control, but also sometimes life-threatening complications, thus constituting a pathology of great urgency.
Conflict of interest: none declared
Financial support: none declared
This work is permanently accessible online free of charge and published under the CC-BY.

Bibliografie
- Avcin T, Cimaz R, Silverman ED, Cervera R, Gattorno M, Garay S, Berkun Y, Sztajnbok FR, Silva CA, Campos LM, Saad-Magalhaes C, Rigante D, Ravelli A, Martini A, Rozman B, Meroni PL. Pediatric antiphospholipid syndrome: clinical and immunologic features of 121 patients in an international registry. Pediatrics. 2008;122:e1100–e1107.
- Corban MT, Duarte-Garcia A, McBane RD, Matteson EL, Lerman LO, Lerman A. Antiphospholipid Syndrome: Role of Vascular Endothelial Cells and Implications for Risk Stratification and Targeted Therapeutics. J Am Coll Cardiol. 2017;69:2317–2330.
- Linnemann B. Antiphospholipid syndrome – an update. Vase. 2018;47:451–464.
- Yelnik CM, Urbanski G, Drumez E, Sobanski V, Maillard H, Lanteri A, Morell-Dubois S, Caron C, Dubucquoi S, Launay D, Duhamel A, Hachulla E, Hatron PY, Lambert M. Persistent triple antiphospholipid antibody positivity as a strong risk factor of first thrombosis, in a long-term follow-up study of patients without history of thrombosis or obstetrical morbidity. Lupus. 2017;26:163–169.
- Ma J, Song H, Wei M, He Y. Clinical characteristics and thrombosis outcomes of paediatric antiphospholipid syndrome: analysis of 58 patients. Clin Rheumatol. 2018;37:1295–1303.
- Meroni PL, Argolini LM, Pontikaki I. What is known about pediatric antiphospholipid syndrome? Expert Rev Hematol. 2016;9:977–985.
- Berman H, Rodríguez-Pintó I, Cervera R, Gregory S, de Meis E, Rodrigues CE, Aikawa NE, de Carvalho JF, Springer J, Niedzwiecki M, Espinosa G. Catastrophic Registry Project Group (European Forum on Antiphospholipid Antibodies) Pediatric catastrophic antiphospholipid syndrome: descriptive analysis of 45 patients from the “CAPS Registry”. Autoimmun Rev. 2014;13:157–162.
- Nageswara Rao AA, Elwood K, Kaur D, Warad DM, Rodriguez V. A retrospective review of pediatric antiphospholipid syndrome and thrombosis outcomes. Blood Coagul Fibrinolysis. 2017;28:205–210.
- Groot N, de Graeff N, Avcin T, Bader-Meunier B, Dolezalova P, Feldman B, Kenet G, Koné-Paut I, Lahdenne P, Marks SD, McCann L, Pilkington CA, Ravelli A, van Royen-Kerkhof A, Uziel Y, Vastert SJ, Wulffraat NM, Ozen S, Brogan P, Kamphuis S, Beresford MW. European evidence-based recommendations for diagnosis and treatment of paediatric antiphospholipid syndrome: the SHARE initiative. Ann Rheum Dis. 2017;76:1637–1641.
- Madison JA, Zuo Y, Knight JS. Pediatric antiphospholipid syndrome. Eur J Rheumatol. 2020;7(Suppl 1):S3-S12.
- Madison JA, Gockman K, Hoy C, Tambralli A, Zuo Y, Knigh JS. Pediatric antiphospholipid syndrome: clinical features and therapeutic interventions in a single center retrospective case series. Pediatric Rheumatology. 2022;20:17.

Diagnostic accuracy of noninvasive tests for pediatric Helicobacter pylori infection
Luiza Elena Bordei, Doina Anca Pleşca
Dobândirea infecţiei cu Helicobacter pylori se produce precoce în copilărie, infecţia fiind regăsită la peste jumătate din populaţia lumii. Majoritatea pacienţilor pediatrici infectaţi sunt asimptomatici....
Liver involvement in cystic fibrosis in children
Bogdan A. Stana, Laura Bozomitu, Sabina Grigoraş, Paula Popovici
Printre atingerile sistemice din mucoviscidoză se regăseşte şi afectarea hepatobiliară, unele determinări genetice ale bolii manifestându-se cu boală hepatică asociată fibrozei chistice (CFLD). ...
Introduction to the use of artificial intelligence in pediatrics
Tudor Lucian Pop
Inteligenţa artificială (IA) reprezintă un domeniu fascinant, care a captat atenţia oamenilor de ştiinţă şi a cercetătorilor. Definiţiile I...
Utilizarea dispozitivelor electronice în prima copilărie (0-5 ani): beneficii educaționale, riscuri de neurodezvoltare și recomandări clinice bazate pe dovezi – o sinteză narativă
Adriana Mihai, Ileana-Katerina Ioniuc, Oana-Raluca Temneanu, Paula Popovici, Alice-Nicoleta Grudnicki, Alina Murgu
Copiii cu vârsta mai mică de 5 ani cresc imersați în medii digitale bazate pe ecrane într-o măsură fără precedent istoric....
Corpii străini intrabronșici la copii: dificultăți diagnostice și terapeutice
Miruna-Bianca Pîslaru, Paula Popovici, Maria-Alessandra Iuga, Cristina Bânțu, Ioana Cernescu, Alina Murgu, Bogdan-Aurelian Stana, Andreea Chiper, Tania-Elena Rusu
Aspirația de corp străin reprezintă o cauză majoră de obstrucție a căilor respiratorii la pacienții pediatrici, însă diagnosti...