Inversia pericentrică a cromozomului 9: un fenotip anormal
Pericentric inversion of chromosome 9: an abnormal phenotype
Abstract
Pericentric inversion of chromosome 9 is considered a balanced structural anomaly. More often cited as a relatively common part of the normal human karyotype, this chromosomal rearrangement could correlate with infertility, abortions and different abnormal clinical conditions. A 5-month-old infant was admitted to our hospital for poor feeding and loose stools. The physical examination revealed an underweight boy (under the fifth percentile) with plagiocephaly, dysmorphic and low-set ears, hypertelorism, saddle nose and micrognathia. The abnormal phenotype also contained long limbs, arachnodactyly, pilonidal sinus and a systolic ejection murmur in the pulmonic area. He was unresponsive to auditory stimuli. The ENT examination described bilateral sensorineural hearing loss. Based on dysmorphic appearance and karyotyping of peripheral, the case was classified as a pericentric inversion of chromosome 9 (p11q13). Despite being categorized as a clinically insignificant variant to a cytogenetic finding, further research is still needed for the few cases where specific pathologies were associated with pericentric inversion of chromosome 9.Keywords
pericentric inversion of chromosome 9karyotypeabnormal phenotypeRezumat
Inversia pericentrică a cromozomului 9 este considerată o anomalie structurală echilibrată. Frecvent citată ca fiind comună cariotipului uman normal, această rearanjare cromozomială s-ar putea corela cu infertilitate, avorturi şi cu diferite patologii. Prezentăm cazul unui sugar, în vârstă de 5 luni, internat în spitalul nostru pentru dificultăţi de alimentare şi scaune de consistentă redusă. Examenul clinic a evidenţiat un sugar cu greutate redusă (sub percentila 5), cu plagiocefalie, urechi dismorfe şi jos inserate, hipertelorism, baza nasului aplatizată şi cu micrognaţie. Fenotipul anormal mai cuprindea membre lungi, arahnodactilie, sinus pilonidal şi prezenţa unui suflu sistolic în aria pulmonară. Sugarul nu răspundea la stimulii auditivi, iar examenul ORL a descris hipoacuzie neurosenzorială bilaterală. Pe baza aspectului dismorf şi a cariotipului periferic, cazul a fost încadrat ca o inversie pericentrică a cromozomului 9 (p11q13). Deşi este clasificată ca o variantă nesemnificativă clinic, sunt încă necesare cercetări suplimentare pentru puţinele cazuri în care diverse patologii specifice au fost asociate cu inversia pericentrică a cromozomului 9.Cuvinte Cheie
inversia pericentrică a cromozomului 9cariotipfenotip anormalIntroduction
Pericentric inversion of chromosome 9 – inv(9) – is one of the most frequent chromosomal variations(1-3), being described in 1-3% of the general population(1). The prevalence varies among different ethnic groups(3). The minor rearrangement in the heterochromatic region of chromosome 9 has been considered a balanced structural anomaly, most often with no phenotypic impact. Despite being widely discussed, the clinical significance is still uncertain. As a relatively common part of the human karyotype, this heterochromatic variant has been correlated with intellectual disability(4-7), schizophrenia(3,4), growth failure(3-5,7), skeletal, cardiac(3,5,8) and genital malformations(3,5,8,9). Moreover, contradictory studies can be found on the association between pericentric inversion of chromosome 9, infertility(1,3-24) and cancer predisposition(3-5,10,12).
Our paper aims at presenting the case of a 5-month-old male infant with an array of congenital abnormalities. We described the chromosomal rearrangement by karyotyping and the clinical usefulness of the correlation between an abnormal phenotype and pericentric inversion of chromosome 9, considered a balanced variation of the human karyotype. Additionally, we reviewed the literature for developmental delay and dysmorphic features similar to those present in our patient.
Case presentation
A couple of parents presented to our hospital with a 5-month-old male infant for poor feeding and loose stools. The infant was the fourth born to healthy non-consanguineous parents. There was no history of drug intake, radiation or chemical exposure during pregnancy. No previous abortions and no older siblings with abnormal phenotypes were found. The antenatal period was uneventful. The routine ultrasounds reported normal findings during pregnancy, until the third-semester examination depicted oligohydramnios. The male child was delivered at 34 weeks of gestation by caesarean section, weighing 2100 g (under the third percentile), with a length of 47.9 cm (the 15th percentile) and with a head circumference of 32 cm (the third percentile).
The physical examination revealed an underweight boy (6 kg, under the third percentile), average length (66 cm, the 50th percentile), plagiocephaly, microcephaly, dysmorphic and low-set ears, hypertelorism, long philtrum, saddle nose and micrognathia. The abnormal phenotype (Figure 1a and b) included: long limbs, arachnodactyly, a fifth finger clinodactyly, camptodactyly of the fingers I, II and III, “sandal gap” deformity, pilonidal sinus and a systolic ejection murmur in the pulmonic area. Even unresponsive to auditory stimuli, he appeared cheerful and presented mild motor developmental delay.
The laboratory investigations revealed an increased alkaline phosphatase, immune globulin G deficiency and undigested fiber in the stool test. The ENT examination described bilateral sensorineural hearing loss. Moreover, a patent foramen ovale was present on cardiac ultrasound, and renal ultrasound identified mild left hydronephrosis. Lactose intolerance genetic test resulted in 13910 C/T and 22018 G/A heterozygous genotype.
The karyotyping was performed due to the infant’s dysmorphic appearance. After 72-hour PHA-stimulation of peripheral blood lymphocytes and G-banding, the cultures were examined in light microscopy. The cytogenetic analysis was performed on twenty metaphase plaques. The chromosomal imbalances were described according to the International System for Human Cytogenetic Nomenclature (ISCN) 2016. As a result of the karyotyping of peripheral, the case was classified as a pericentric inversion of chromosome 9 (p11q13). Parental karyotype could not be performed due to the father’s refusal of consent, while further investigation of the mother’s karyotype encountered no genomic abnormalities.
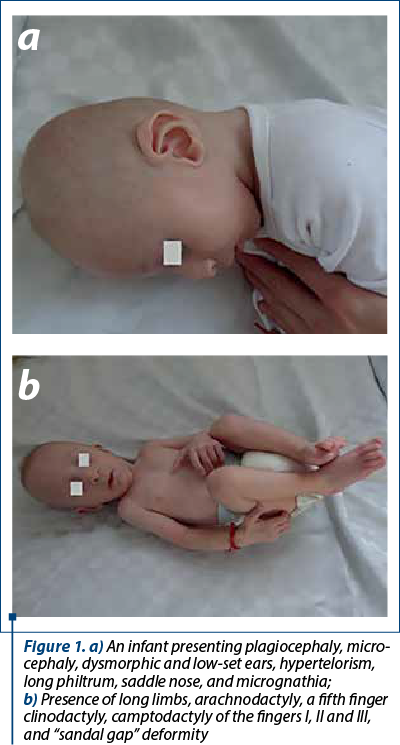
Presentation at 18 months
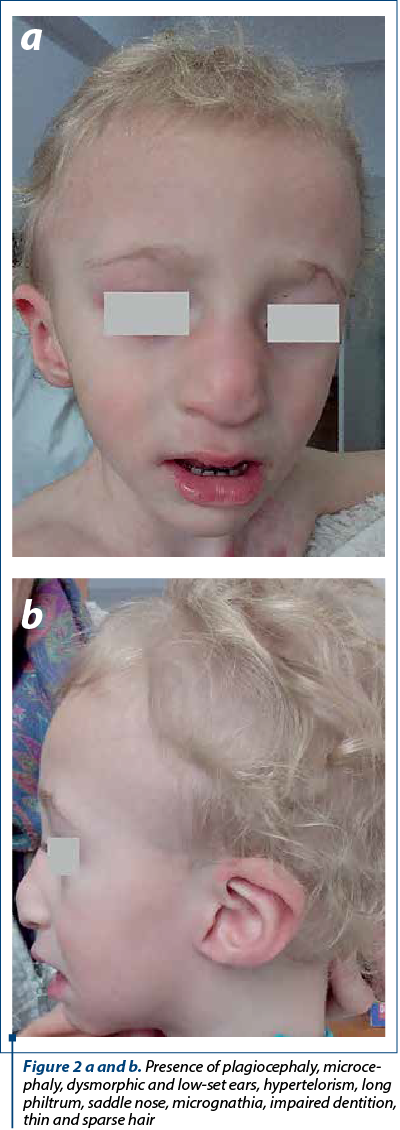
The evaluation at 18 months revealed an underweight boy (9 kg, the third percentile), with tall stature (90 cm, above the 95th percentile), presenting the craniofacial dysmorphisms previously described, associating impaired dentition (delayed tooth eruption and cavities), pectus excavatum, thin and sparse hair (Figure 2a and b). In addition, he appeared cheerful but with moderate neurodevelopmental delay, walking difficulties and with severe speech delay. He suffered a traumatic (hot water chest-burn) anterior chest lesion. The head CT revealed the persistence of cavum septum pellucidum and cavum vergae.
Discussion
Pericentric inversion of chromosome 9 is one of the most common morphological chromosomal variations(1-3). The frequency of inv(9) is variable among different ethnic groups, ranging from a high rate in the African population (3.57%) to a relatively low rate in Caucasians (0.73%)(3). The incidence of this fetal disorder was higher in females than in males, without a specific explanation. Maternal origin was inclined (1.3 times) more often than paternal origin, with only one de novo event being described in the most extensive study on carriers of chromosome 9 variants(12).
Developmental delay and dysmorphic features affect 2-3% of the pediatric population. In a limited number of cases, additional research is recommended for chromosomal imbalances using the G-banded karyotype and chromosomal microarray(25). After discovering the congenital abnormalities, the genetic test result concluded that our case was a minor rearrangement, namely a pericentric inversion of chromosome 9 (p11q13). This unifying diagnosis accounts for a part of the patient’s dysmorphic features, skeletal, cardiac and urological malformations.
Molecular studies suggest that the complex structural organization of the pericentric region of chromosome 9 may contain susceptible breakpoints with a higher potential of generating pericentric inversions, deletions, duplications and other types of variations. The pathogenesis of this disorder involves the heterochromatic region of chromosome 9, leading to a variety of clinical presentations. Among these heterochromatic variants, the pericentric inv(9) is considered a balanced structural anomaly(2,4).
Although firstly described more than four decades ago(26), the clinical significance of chromosome 9 variants is still uncertain. More often cited as a relatively common part of the normal human karyotype, this chromosomal rearrangement could be associated with dysmorphic features: growth failure(3-5,7), synophrys, low hairline(5), microcephaly(3,5) and micrognathia(5).
A few studies have drawn the correlation between inv(9) and various anomalies such as intellectual disability(4-7), schizophrenia(3,4), bipolar disorder(4), skeletal, cardiac(3,5,8) and genital malformations(3,5,8,9). The heterochromatin polymorphism of chromosome 9 has been mentioned in patients with hematological malignancies(3-5,10,12). It is still uncertain whether inv(9) and malignant diseases could be regarded as a familial inheritance or due to acquired conditions(10).
Previous literature has mentioned the association between structural chromosomal anomalies and reproductive failure(1,3-24). A prospective study carried out over more than fifteen years in 895 individuals with a history of infertility observed that the most common abnormal karyotype contained the pericentric inversion of chromosome 9(11). Some authors report the heterochromatin variants and underlying reproductive failure with a higher incidence in females(11,13), while others describe a frequent correlation between inv(9) and azoospermia(14,15). We found the malformations mentioned before in our present case, but other clinical conditions could still develop.
We encountered various similarities regarding the phenotypic anomalies between our patient and a case of pericentric inversion of chromosome 9 and a de novo 6-Mb 9 (p13.1p11.2) duplication reported by Malinverni et al.(7): hypertelorism, low set ears, long philtrum, dysmorphic nose, “sandal gap” deformity, a fifth finger clinodactyly, neurodevelopmental and growth delay. The aforementioned study described the father’s karyotype with the same positive C-banding as the child(7).
One study analyzed the frequency of chromosomal polymorphisms among high-risk cases by performing maternal serum screening followed by amniocentesis. Inv(9) was the second most common polymorphic variant, specifically inv(9) (p11q13). In almost half of the parents who underwent chromosomal karyotype analysis, it was shown that fetal inv(9) was inherited from one parent and did not present an abnormal clinical phenotype. In this regard, the authors mentioned that inv(9) could be associated with abnormal clinical phenotype only if pregnant couples present other disorders that can lead to infertility or to an unfavorable pregnancy outcome(24).
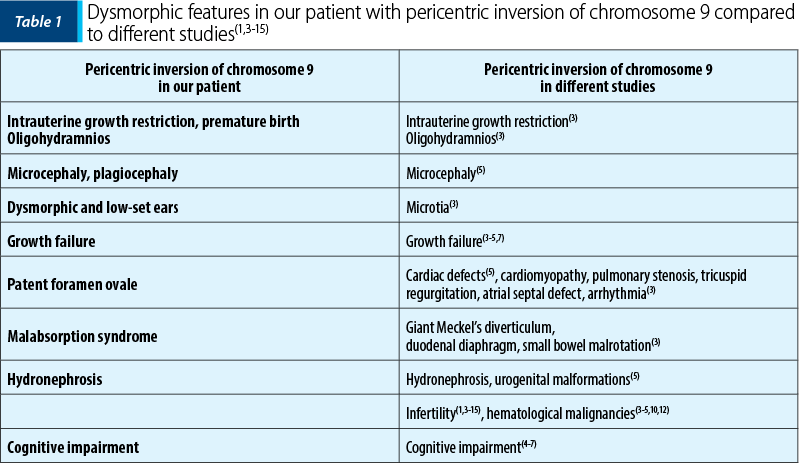
There are various similarities between our present case and the various malformations associated with de novo inv(9) mentioned in a study in 431 neonates with congenital disorders(3). Similar malformations were also described in another study which listed 157 cases of inv(9), with inversions inherited from one of the parents(4). A significant key point in our complete diagnosis would have been the chromosomal analysis of both parents. We were able to investigate only the mother’s karyotype, which encountered no genomic abnormalities, while the father’s consent was not obtained. Parental chromosomal analysis is essential to assess the origin of the rearrangement to offer more accurate genetic counseling. We emphasized the clinical features of our case compared to other studies regarding the dysmorphic anomalies of the heterochromatic region of chromosome 9 (Table 1).
Numerous studies have described developmental delay and dysmorphic features similar to those present in our patient. Marfan syndrome is a rare disorder diagnosed in infants, although a few skeletal findings may be encountered from birth, including arachnodactyly, dolichocephaly, long bones and joint laxity, micrognathia and pectus deformity. A broad range of malformations complete this syndrome, ranging from cardiac and aortic disease to ocular, pulmonary and skin abnormalities(27). VACTERL association is a disorder that often includes vertebral defects, digestive tract and cardiac defects, renal and limbs malformations(28). Congenital craniofacial anomalies are connected to defects associated with neural crest cells. Treacher Collins syndrome is a severe congenital disorder that implies craniofacial malformations, such as cleft palate, hypoplastic facial bones, malformation of the external and middle ear(29). The chromosome 17q21.31 deletion syndrome, also known as Koolen-De Vries syndrome, is characterized by particular facial features, including long face and philtrum, bulbous nasal tip, spaced teeth and dysmorphic ears. Other relevant component manifestations are growth failure, intellectual disability, hypotonia and friendly behavior. Furthermore, epilepsy, heart defects and kidney anomalies are attributed to subjects with this chromosome deletion and KANSL1 mutation(30).
Conclusions
This case highlights the combination of an abnormal phenotype and one of the most common balanced structural anomalies. Despite being categorized as a clinically insignificant variant to a cytogenetic finding, further research is still needed for the few cases where specific pathologies were associated with pericentric inversion of chromosome 9. Additional investigations of the breakpoint region could provide a better understanding of how to differentiate a common heteromorphism from an abnormal aberration. More awareness should be given towards various studies that continue to draw a correlation between this chromosomal inversion and an array of abnormalities.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Abdalla EM, El-Kharadly RN. Pericentric Inversion of Chromosome 9 in a Consanguineous Couple with Molar Pregnancies and Spontaneous Abortions. Laboratory Medicine. 2012;43(5):212–216.
-
Wang JC, Boyar FZ. Chromosomal microarray analysis as the first-tier test for the identification of pathogenic copy number variants in chromosome 9 pericentric regions and its challenge. Molecular Cytogenetics. 2016;9(64).
-
Jeong SY, Kim BY, Yu JE. De Novo Pericentric Inversion of Chromosome 9 in Congenital Anomaly. Yonsei Med J. 2010;51(5):775-80.
-
Demirhan O, Pazarbasi A, Suleymanova-Karahan D. Correlation of Clinical Phenotype with a Pericentric Inversion of Chromosome 9 and Genetic Counseling. Saudi Med J. 2008;29(7):946-951.
-
Akbas E, Senli H, Hallioglu O. Association of Pericentric Inversion of Chromosome 9 (inv[9] [p11q13]) and Genetic Diseases: Case Report. Laboratory Medicine. 2010;41(7):439.
-
Wyandt HE, Wilson GN, Tonk VS. Human Chromosome Variation: Heteromorphism, Polymorphism and Pathogenesis, 2nd ed.; Springer Singapore, 2017, pp 13-36.
-
Malinverni ACM, Colovati ME, Perez ABA. Unusual Duplication in the Pericentromeric Region of Chromosome 9 in a Patient with Phenotypic Alterations. Cytogenet Genome Res. 2016;150(2):100-105.
-
Le LT, Dinh N, Hoang M, Tran NT, An LT, Vu QD, Ngo ND, Le HT. AB015. Study on pericentric inversion of chromosome 9 and congenital abnormalities in children. Annals of Translational Medicine. 2017;5(Suppl 2):AB015. doi:10.21037/atm.2017.s015.
-
Arnold A. Primary hyperparathyroidism: molecular genetic insights and clinical implications. Endocrine Abstracts. 2017;50 PL1.
-
Vijay S, Narayanan G, Sarojam S. Enigmatic Inv(9): A Case Report on Rare Findings in Hematological Malignancies. Iran Red Crescent Med J. 2016;18(4):25062.
-
Demirhan O, Tanrıverdi N, Süleymanova D. Chromosomal Analysis of Couples with Bad Obstetric History. J Clin Dev Biol. 2016;1:3.
-
Kosyakova N, Grigorian A, Liehr T. Heteromorphic variants of chromosome 9. Molecular Cytogenetics. 2013;6(14).
-
Šípek AJ, Panczak A, Mihalová R. Pericentric Inversion of Human Chromosome 9 Epidemiology Study in Czech Males and Females. Folia Biologica (Praha). 2015;61:140-146.
-
Mierla D, Jardan D, Stoian V. Chromosomal Abnormality in Men with Impaired Spermatogenesis. Int J Fertil Steril. 2014;8(1):35-42.
-
Pal AK, Ambulkar PS, Sontakke BR. Role of nuclear and mitochondrial genes in human male infertility: a review. Nucleus. 2017;60:209–220.
-
Houmaid H, El Bekkay C, Nassereddine S, Talbi H, Amehdare L, Hilali A. Chromosomal Abnormalities in 238 Couples with Recurrent Miscarriages in Morocco. Open Journal of Genetics. 2018;8:15-22.
-
Pal AK, Ambulkar PS, Waghmare JE, Wankhede V, Shende MR, Tarnekar AM. Chromosomal Aberrations in Couples with Pregnancy Loss: A Retrospective Study. J Hum Reprod Sci. 2018;11(3):247-253. doi: 10.4103/jhrs.JHRS_124_17.
-
Değirmencia B, Solakb M, Yildiz SH, Erdogan MO, Elmas M, Fistik T. Evaluation of cytogenetic and y chromosome microdeletion analyzes in infertile cases. Meta Gene. 2019;19:78-81.
-
Fan Q, Zhang J, Cui Y, Wang C, Xie Y, Wang Q, Wu L. The synergic effects of CTLA-4/Foxp3-related genotypes and chromosomal aberrations on the risk of recurrent spontaneous abortion among a Chinese Han population. Journal of Human Genetics. 2018;63:579–587.
-
Inan C, Sayin NC, Dolgun ZN, Gurkan H, Erzincan SG, Uzun I, Sutcu H, Ates S, Atli E, Varol F. Prenatal diagnosis of chromosomal polymorphisms: most commonly observed polymorphism on Chromosome 9 have associations with low PAPP-A values. J Matern Fetal Neonatal Med. 2019, 32(10):1688-1695. doi: 10.1080/14767058.2017.1416079.1-8.
-
Liang S, Yang J, Wu H, Teng X, Duan T. Effects of chromosome 9 inversion on IVF/ICSI: A 7-year retrospective cohort study. Mol Genet Genomic Med. 2019;7(9):e856. doi: 10.1002/mgg3.856.
-
Nonaka T, Takahashi M, Nonaka C, Enomoto T, Takakuwa K. The analysis of chromosomal abnormalities in patients with recurrent pregnancy loss, focusing on the prognosis of patients with inversion of chromosome (9). Reprod Med Biol. 2019;18(3):296-301. doi: 10.1002/rmb2.12281.
-
Xie X, Li F, Tan W, Tang J. Analysis of the clinical features of pericentric inversion of chromosome 9. Journal of International Medical Research. 2020;48(9):1-9.
-
Rawal L, Kumar S, Mishra SR, Lal V, Bhattacharya SK. Clinical Manifestations of Chromosomal Anomalies and Polymorphic Variations in Patients Suffering from Reproductive Failure. J Hum Reprod Sci. 2020;13(3):209-215. doi: 10.4103/jhrs.JHRS_46_19.
-
Gupta SN, Gupta VS, Ahmed A. “Common Developmental Delay”. In: Full-term Children: A Common Neurological Profile to Aid in Clinical Diagnosis. J Clin Dev Biol. 2016;1(2):1-8.
-
Wahrman J, Atidia J, Goitein R. Pericentric inversions of chromosome 9 in two families. Cytogenetics. 1972;11:132-144.
-
Reyes-Hernández OD, Palacios-Reyes C, Chávez-Ocaña S. Skeletal manifestations of Marfan syndrome associated to heterozygous R2726W FBN1 variant: sibling case report and literature review. BMC Musculoskeletal Disorders. 2016;17(79).
-
Chen Y, Liu Z, Chen J. The genetic landscape and clinical implications of vertebral anomalies in VACTERL association. J Med Genet. 2016;53(7):431-7.
-
Sakai D, Dixon J, Achilleos A. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat Commun. 2016;7:10328.
-
Zollino M, Marangi G, Ponzi E. Intragenic KANSL1 mutations and chromosome 17q21.31 deletions: broadening the clinical spectrum and genotype–phenotype correlations in a large cohort of patients. J Med Genet. 2015;52(12):804-14.

Schimbare de paradigmă în MIS-C – cazul unei furtuni perfecte
Alina-Costina Luca, Ioana-Alexandra Pădureţ, Magdalena Starcea, Elena Macsim, Cristina Alexa, Alexandru David, Bogdan A. Stana
Infarctul renal acut la copii şi adolescenţi este un eveniment rar, asociat de obicei cu anomalii cardiace, cu status hipercoagulant, boli cu potenţial trombotic local, infecţii sistemice sau cu ...
Purpura fulminans, modalitate de debut clinic în sindromul inflamator multisistemic la copil (MIS-C)
Bianca Raluca Mariş, Alina Grama, Simona Căinap, Alexandra Mititelu, Georgiana Laura Cioancă, Gabriel Benţa, Claudia Sîrbe, Alexandra Mariş, Laura Bodea, Tudor Lucian Pop
Încă de la începutul anului 2020, boala coronavirus 2019 (COVID-19) a reprezentat o mare problemă de sănătate pe întreg globul, m...
Impactul esofagitei eozinofilice asupra calităţii vieţii la populaţia pediatrică
Ioana Maria Otilia Lică, Iulia Florentina Ţincu, Doina Anca Pleşca
Esofagita eozinofilică, deşi relativ recent introdusă drept concept în literatura de specialitate şi în rândul afecţiunilor digestive, a evoluat cu o incidenţă din ce în ce mai mare în rândul populaţiei pediatrice. ...
Schimbare de paradigmă în MIS-C – cazul unei furtuni perfecte
Alina-Costina Luca, Ioana-Alexandra Pădureţ, Magdalena Starcea, Elena Macsim, Cristina Alexa, Alexandru David, Bogdan A. Stana
Infarctul renal acut la copii şi adolescenţi este un eveniment rar, asociat de obicei cu anomalii cardiace, cu status hipercoagulant, boli cu potenţial trombotic local, infecţii sistemice sau cu ...
Purpura fulminans, modalitate de debut clinic în sindromul inflamator multisistemic la copil (MIS-C)
Bianca Raluca Mariş, Alina Grama, Simona Căinap, Alexandra Mititelu, Georgiana Laura Cioancă, Gabriel Benţa, Claudia Sîrbe, Alexandra Mariş, Laura Bodea, Tudor Lucian Pop
Încă de la începutul anului 2020, boala coronavirus 2019 (COVID-19) a reprezentat o mare problemă de sănătate pe întreg globul, m...