Sindromul de regresie caudală – afecţiune congenitală rară cu implicaţii sistemice multiple
Caudal regression syndrome – a rare congenital disorder with multiple systemic implications
Abstract
Background. Caudal regression syndrome is a rare congenital disorder (frequency: 1-2.5 cases/100,000 births) of unknown cause. It consists in a developmental defect of the sacred-lumbar area, associated with a large spectrum of craniofacial, genitourinary and gastrointestinal anomalies. Most cases were encountered in children from insulin-dependent mothers. Case presentation. We report the case of a 1-year-and-2-month-old boy admitted in the Pediatrics Department of the “Grigore Alexandrescu” Emergency Clinical Hospital for Children, Bucharest, for failure to thrive and recurrent urinary tract infections. From his medical past, we mention: lumbar myelomeningocele partially closed through in utero surgery, sacral agenesis, hydrocephalus with ventriculoperitoneal shunt set up at 1 month, nephrocalcinosis. Amniocentesis identified a normal karyotype. The clinical examination revealed altered auxologic parameters, global hypotonia, pelvic girdle muscles and lower limbs muscles hypotrophy, along with clubfeet. The laboratory tests showed normal liver and renal functions; vitamin D3, serum aldosterone and renin levels were normal. Upper gastrointestinal tract barium with X-ray revealed massive gastroesophageal reflux. The abdominal ultrasound revealed bilateral nephrocalcinosis without any other structural kidney anomalies. MRI confirmed the sacral agenesis. The genetic advice established the diagnosis of caudal regression syndrome; mucopolysaccharidosis type IV was ruled out due to normal enzymatic testing. Considering the severe failure to thrive in a child with plurimalformative disorder, the surgical therapy consisting in gastrostomy combined with Nissen fundoplication was recommended. Conclusions. Caudal regression syndrome is a rare congenital anomaly with numerous systemic consequences which takes combined efforts from a multidisciplinary team and the family in order to proper monitor the child and find the best therapeutic approach.Keywords
sacral agenesiscaudal regressionmalnutritionRezumat
Introducere. Sindromul de regresie caudală reprezintă o afecţiune congenitală rară (1-2,5 cazuri/100.000 de naşteri) cu etiologie necunoscută şi constă într-un defect de dezvoltare a regiunii lombosacrate asociat cu multiple malformaţii craniofaciale, genitourinare şi gastrointestinale. Cele mai multe cazuri au fost raportate la copiii proveniţi din mame cu diabet zaharat insulino-necesitant. Prezentare de caz. Raportăm cazul unui băiat în vârstă de 1 an şi 2 luni, internat în Departamentul de pediatrie al Spitalului Clinic de Urgenţe pentru Copii „Grigore Alexandrescu”, Bucureşti, pentru falimentul creşterii şi infecţii urinare recurente. Din istoric reţinem: mielomeningocel lombar parţial închis in utero, agenezie sacrală, hidrocefalie cu şunt ventriculoperitoneal montat la vârsta de o lună şi nefrocalcinoză. Amniocenteza a identificat un cariotip convenţional. Examenul clinic a decelat hipotrofie staturoponderală marcantă, hipotonie generalizată, hipotrofia musculaturii centurii pelviene şi a membrelor inferioare şi calcaneovalgus. Investigaţiile de laborator au evidenţiat funcţie hepatică şi funcţie renală normale şi 25-OH vitamina D3, aldosteron şi renina serică în limite normale. Tranzitul baritat a decelat reflux gastroesofagian masiv. Ecografia abdominală a descris prezenţa ambilor rinichi şi nefrocalcinoză bilaterală. RMN-ul a confirmat absenţa sacrului. Consultul genetic a stabilit diagnosticul de sindrom de regresie caudală. A fost exclusă o mucopolizaharidoză de tip IV, testarea enzimatică fiind negativă. Având în vedere malnutriţia severă la un copil cu multiple malformaţii, s-au propus pentru optimizarea curbei de creştere efectuarea fundoplicaturii Nissen şi montarea de gastrostomă. Concluzii. Sindromul regresiei caudale este o afecţiune congenitală rară, cu multiple implicaţii sistemice, ce necesită eforturi combinate din partea unei echipe multidisciplinare şi a familiei pentru monitorizare şi stabilirea unui protocol terapeutic optim.Cuvinte Cheie
agenezie sacralăregresie caudalămalnutriţieIntroduction
Caudal regression syndrome (CRS) – also known as caudal dysplasia, sacral agenesis or sacral defect with anterior meningocele – is a rare congenital condition of unknown cause which consists in a developmental defect of the lumbosacral area. It is associated with craniofacial malformations (cleft palate), renal anomalies (horseshoe kidney, unilateral renal agenesis, ureteral duplications, vesicoureteral reflux, neurogenic bladder), genital anomalies (hypospadias, cryptorchidism, rectovaginal fistula) and gastrointestinal malformations (anal imperforation)(1,2).
Case presentation
We present the case of a 1-year-and-2-month-old boy admitted in the Pediatrics Department of the “Grigore Alexandrescu” Emergency Clinical Hospital for Children, Bucharest, in September 2019, for failure to thrive, food refusal and recurrent urinary tract infections.
From his medical past, it is important to mention that he was prematurely born at 36 weeks of gestation through caesarean section, with a regular Apgar score of 8. He was diagnosed in utero (19 weeks) with myelomeningocele, which was partially closed at 26 weeks of gestation and completely resolved three days after birth. Considering the antenatal diagnosis of myelomeningocele, amniocentesis was highly recommended and revealed a conventional male karyotype 46 XY.
Thoraco-lumbar X-ray of the spine described absent sacrum, butterfly-shaped T6 vertebrae, and the magnetic resonance imaging confirmed sacral agenesis and identified cerebellum hypoplasia with significant ventriculomegaly. A ventriculoperitoneal shunt was set up at 1 month, with good outcome.
An abdominal ultrasound performed one week after birth revealed bilateral nephrocalcinosis, without any other structural kidney anomalies. Since 2 months old, the child experienced numerous urinary tract infections (with Klebsiella pneumoniae and Enterococcus faecalis). At 10 months old, a nephrologist established the diagnoses of nephrocalcinosis, giant bladder and possible tubular renal acidosis type IV, which was ruled out due to normal serum aldosterone and renin values.
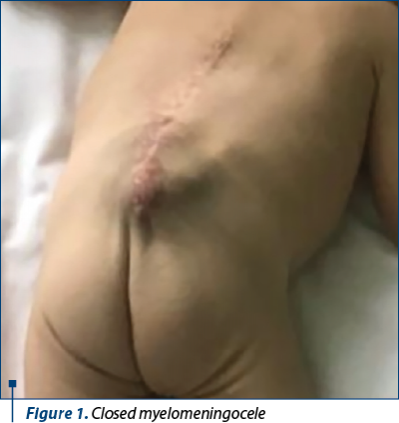
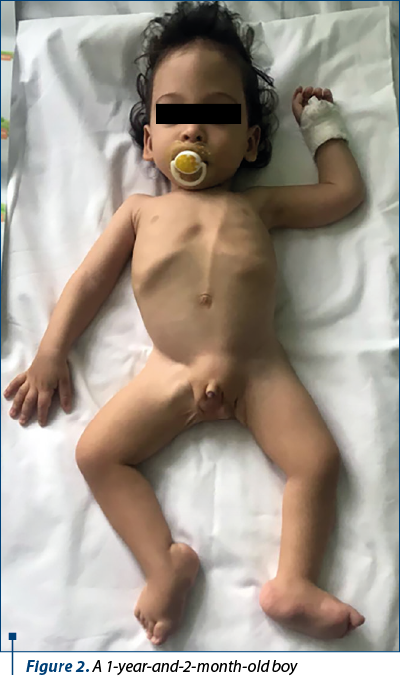
The relevant information regarding his family medical history includes overweight mother with eventual metabolic syndrome; the father and the older sister (13 years old) were both healthy.
The clinical examination encountered a child with a severely altered auxologic parameters, weight = 5300 g (below percentile 0.1), height = 65 cm (below percentile 0.6), head circumference = 44 cm (percentile 0.6), with pale skin, scraggy look, global hypotonia, pelvic girdle and lower limbs muscles hypotrophy, bilateral clubfeet and penile hypoplasia, functional ventriculoperitoneal shunt without signs of intracranial hypertension, apparently without sight or hearing disfunctions.
The laboratory tests revealed proper liver and renal functions, regular fat and protein metabolism levels, with normal C reactive protein and urinalysis. Serum aldosterone and renin values were normal, thus excluding a possible renal tubular acidosis type IV.
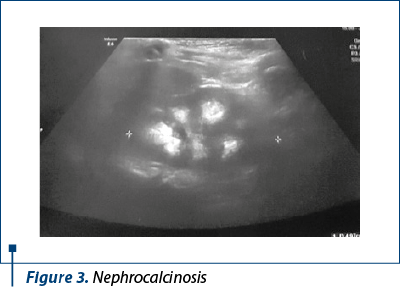
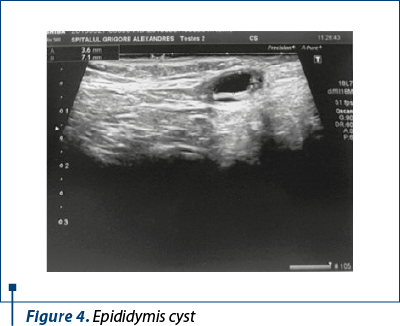
The abdominal ultrasound described bilateral renal non-homogenous calyceal images, considered to be nephrocalcinosis (Figure 3). Pelvic sonography encountered right and left epididymis cysts, measuring 7.1/3 and 2/1.7, without clinical consequences (Figure 4).
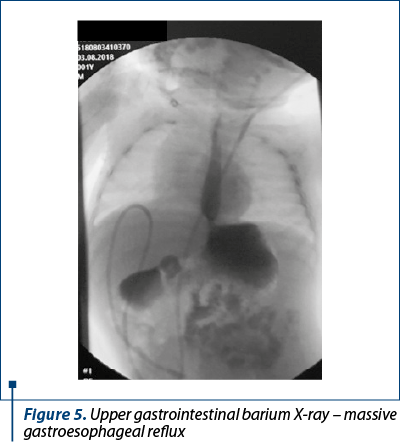
Because weight balance was unsatisfactory and the clinical signs suggested gastroesophageal reflux disease, we decided to perform an upper gastrointestinal barium X-ray, which described significant gastroesophageal reflux in dorsal decubitus position (Figure 5).
In order to establish the right diagnosis, the child underwent a neurological examination which confirmed the global hypotonia with lower limbs muscle weakness and motor deficiency. The neurosurgical examination described a functional ventriculoperitoneal shunt and mostly right-sided paraparesis, without signs of intracranial hypertension. Echocardiography didn’t find any structural cardiac anomalies. Also, orthopedic examination described the sacrolumbar defect, bilateral clubfeet, left hip displasia and both-sided talus valgus.
Keeping in mind that this is a child with congenital disorder, the next logical step was the genetic advice, which confirmed the diagnosis of caudal regression syndrome on grounds of clinical and imaging criteria (sacral agenesis, myelomeningocele), normal karyotype. The conditions with similar features associating skeletal defects and weight-stature delays have been taken into account: Currarino triad (we didn’t encountered anorectal malformations), VACTERL anomaly (we didn’t find cardiac, gastrointestinal or renal abnormalities), mucopolysaccharidose type IV (the ultrasound examination didn’t describe hepatosplenomegaly or cardiac malformations, the enzymatic testing was normal).
Due to the severe failure to thrive and feeding disorder in a child with complex malformative pathology, the surgical assesment consisting in Nissen fundoplication combined with gastrostomy was highly recommended in order to improve the weight balance. The family did not go along with surgical therapy because of its long-term implications.
Caudal regression syndrome type 3 (myelomeningocele with ventriculoperitoneal shunt, sacral agenesis), associated with severe malnutrition and neuromotor development delays, requires a multidisciplinary assesment, keeping in mind the severe prognosis due to renal complications, but also the orthopedic defects which can lead to walking impairment.
Even though the child had an early diagnosis of sacral agenesis and myelomeningocele, the caudal regression syndrome was a late discovery, starting with feeding impairment, severe failure to thrive and recurrent urinary tract infections.
The patient continues to be monitored in our pediatrics department and he will receive prophylactic antibiotic therapy in order to prevent further urinary tract infections; he will undergo periodic urine analysis and renal ultrasound evaluation, considering that children with sacral agenesis may develop in time neurogenic bladder and chronic kidney disease. Neuromotor and neuropsychiatric development should be evaluated from time to time, although patients with caudal regression have an intellect within normal limits. Musculoskeletal impairment should be monitored by an orthopedist and should benefit from physical therapy.
Discussion
Caudal regression syndrome is a rare congenital condition which affects the sacred-lumbar area, both the bones and spinal cord, with a reported incidence ranging from 1 to 2.5 cases/100,000 births, but it has a much higher occurence rate (200-400 higher) in children from insulin-dependent diabetic mothers(1,3).
The causes of this anomaly remain unknown, being more likely the result of the interaction between multiple genetic and environmental factors which appear to act during weeks 3-7 of gestation. The only acknowledged risk factor associated with caudal regression is maternal diabetes (approximately 20% of mothers who give birth to children with sacral agenesis have insulin-dependent diabetes), although the pathogenic mechanism was not yet identified(1,2,4). The patient’s mother is overweight, with eventual metabolic syndrome, but she has not been diagnosed with diabetes.
Like many other congenital disorders, caudal regression syndrome can be diagnosed before birth with transabdominal ultrasound, which identifies eloquent abnormalities such as diminished cranio-caudal length (in the first trimester) and the absence of sacrum, along with hypoplastic iliac blades or “frog-like” appearance of the fetus, also known as “Buddha’s pose” (in the second trimester). In most cases, the antenatal diagnosis is established late, after 20-22 weeks of gestation(3,5). Myelomeningocele was described in our patient on abdominal ultrasound at 19 weeks of gestation, but the classic sonographic signs of caudal regression syndrome were not found.
Magnetic nuclear resonance (MRI) is the next noninvasive imaging investigation which has a major role in the diagnosis of caudal dysplasia. It is useful both before and after birth, being also a necessary tool because it confirms the sacral absence, spinal cord lesions and possible associated anomalies(5,6). A spine MRI was performed in our case and found sacrococcygeal agenesis and closed myelomeningocele.
The diagnosis of caudal regression syndrome is based on clinical and imaging data, such as absent sacrum, myelomeningocele and the consequences of these anomalies – small hip bones with limited capacity of movement, hypoplastic gluteal muscle, congenital in-ward and up-ward turning feet. Because most cases of caudal dysplasia have a sporadic occurrence and appear in individuals with no family history, the genetic testing is not a fundamental requirement(1,4,5,7). In our case, the genetic advice established the diagnosis of caudal dysplasia based on clinical and imaging criteria; according to literature data, our patient presented with type 3 caudal regression syndrome.
Based on the type of defect, Renshaw classified sacral agenesis in five categories: types 1 and 2 – partial sacral agenesis with good outcome (they require orthopedic therapy); type 3 – total sacral agenesis; type 4 – fusion of soft tissues in both lower limbs; and type 5 – sirenomelia, also known as mermaid syndrome (single femur or tibia), incompatible with life. Types 3-5 are severe forms of caudal dysplasia, associating multiple malformations, such as renal agenesis, horseshoe kidney and lower limbs abnormalities. They have a severe prognosis due to renal and gastrointestinal complications(5,8).
It is important to differentiate caudal regression from other similar pathologies, such as Currarino triad or VACTERL anomaly. Currarino triad is an extremely rare condition, with an autosomal dominant inheritance pattern, in which we encounter partial/complete absence of sacrum, anterior meningocele and anorectal malformations. The cause of Currarino triad is a genetic mutation in the HLXB9 gene. VACTERL is the acronym for a cumulation of congenital defects – vertebral abnormalities, anal atresia, cardiac malformations, tracheal anomalies, esophageal atresia, renal and radial abnormalities, limb defects; the intellect is usually unaffected, such as in children with caudal regression(1,7,9).
Regarding the therapeutic methods, the children with caudal regression syndrome should benefit from a multidisciplinary medical team, including nephrologist, orthopedist, gastroenterologist, nutritional adviser, cardiologist and neurosurgeon(7). Our patient benefited from in utero surgical assessment of myelomeningocele, which was partially closed at 26 weeks of gestation, but developed hydrocephalus and required ventriculoperitoneal shunt.
The majority of patients with caudal dysplasia have different types of lower urinary neurogenic dysfunctions, such as detrusor dyssynergia or detrusor overactivity (both forms of neurogenic bladder). They are more likely to develop chronic kidney disease than those who suffer from neurogenic bladder due to other forms of spina bifida. Anatomic abnormalities of the reno-urinary system can be encountered: vesicoureteral reflux (VUR), hydronephrosis or renal agenesis(7,10,11). In our patient, the abdominal ultrasound described both kidneys, without any structural anomalies, but with bilateral nephrocalcinosis, still of unknown cause.
The medical treatment includes prophylactic antibiotherapy for urinary tract infections, but also anticholinergic agents and alpha-adrenergic antagonists combined with noninvasive electric stimulation of the bladder(11). A study conducted by Cirovic et al., between 2003 and 2008, regarding electrovesical stimulation, which included 49 children with vesical disfunction due to different types of spina bifida, concluded that this new therapeutic approach can significantly improve the urinary parameters and the specific symptoms – nocturnal enuresis, incontinence(12).
Caudal regression syndrome is a disabling condition and, despite an almost normal intellect, the patient has a poor quality of life. But there is still hope for these children – a medical case published in 2017 described the benefits of growth hormone therapy. After five years of treatment, the patient gained full control of bladder sphincter and was able to maintain the independent standing, without any reported side effects(13).
Conclusions
Caudal regression syndrome is a rare congenital condition of unknown cause with multiple systemic implications, which requires multidisciplinary care. A proper assessment of diabetes in the preconceptional period alongside a well-performed antenatal ultrasound screening (9-20 weeks of gestation) have a major role in preventing this disorder. It is also a disabling condition, thus patients are having a poor quality of life despite an intellect within normal range. The prognosis is severe in most cases, especially if renal or digestive complications occur.
Bibliografie
-
Semba K, Yamamura K. Etiology of Caudal Regression Syndrome. Human Genet Embryol. 2013;3:107.
-
Chan BW, Chan KS, Koide T, Yeung SM, Leung MBW, Copp AJ, Loeken MR, Shiroishi T, Shum ASW. Maternal Diabetes Increases the Risk of Caudal Regression Caused by Retinoic Acid. American Diabetes Association. 2002;51(9):2811-2816.
-
Bhatt S, Tandon A, Singh AK, Manchanda S, Jain S, Meena N. Caudal Regression Syndrome: A Case Study With Associated Review of Common Differential Diagnoses Made With Antenatal Sonography. Journal of Diagnostic Medical Sonography. 2017;33(2):130–133.
-
Genetic and Rare Disease Information Center. Caudal regression sequence. Available at: https://rarediseases.info.nih.gov/diseases/6007/caudal-regression-sequence
-
Bouchahda H, Mhabrech HE, Hamouda H, Ghanmi S, Bouchahda R, Soua H. Prenatal diagnosis of caudal regression syndrome and omphalocele in a fetus of a diabetic mother. Pan Afr Med J. 2017;27:128.
-
Iqbal S, Weerakkody Y. Caudal regression syndrome. Radiopaedia. Available at: https://radiopaedia.org/articles/caudal-regression-syndrome
-
Rare Disease Database. Caudal Rgression Syndrome. Available at: https://rarediseases.org/rare-diseases/caudal-regression-syndrome/
-
Sharma S, Sharma V, Awasthi B, Sehgal M, Singla DA. Sacral Agenesis with Neurogenic Bladder Dysfunction – A Case Report and Review of the Literature. JCDR. 2015;9(6):RD08-RD09.
-
Kanagasabai K, Bhat V, Pramod GK, Patil SJ, Kiranmayi S. Severe caudal regression syndrome with overlapping features of VACTERL complex: antenatal detection and follow up. BJR Case Rep. 2016 Dec 8;3(2):20150356.
-
Edrees BM. Caudal Regression Syndrome/neurogenic bladder presented as recurrent urinary tract infection. AJM. 2016;53(3):289-297.
-
Kalicka K, Zajaczkiwska MM, Piechuta L, Czyz J, Kasza A, Majewski M, Wojciechowska M, Sikora P. Neurogenic bladder as a symptom of caudal regression syndrome. Pediatria Polska. 2016;91(6):628-631.
-
Cirović D, Petronić I, Nikolić D, Brdar R, Pavicevic P, Knezevic T. Effects of electrotherapy in treatment of neurogenic bladder in children with occult spinal dysraphism. Srp Arh Celok Lek. 2009;137(9-10):502-5.
-
Devesa J. Growth Hormone and Rehabilitation Promoted Distal Innervation in a Child Affected by Caudal Regression Syndrome – Beyond The Abstract. Available at: https://www.urotoday.com/beyond-the-abstracts/pelvic-health-reconstruction/neurogenic-bladder/96757-growth-hormone-and-rehabilitation-promoted-distal-innervation-in-a-child-affected-by-caudal-regression-syndrome-beyond-the-abstract

Intoleranţa ereditară la fructoză: capcană de diagnostic la sugar?
Alina Grama, Claudia Sîrbe, Alexandra Mariş, Bogdan Bulata, Mariela Militaru, Tudor Lucian Pop
Hereditary fructose intolerance (HFI) – also called fructose-1-phosphate aldolase deficiency – is a rare inherited metabolic disorder characterized by the body’s inability to metabolize fructose or its precursors (sugar,...
Studiu prospectiv privind managementul durerii acute la nou-născutul cu afecţiuni chirurgicale prin corelaţia scalei de durere CRIES cu protocolul terapeutic etapizat
Valentin Munteanu, Ioana Alecsandra Munteanu, Iulia Ciongradi, Ioan Sârbu, Elena Hanganu, Bogdan A. Stana, Maria Stamatin
The last 2-3 decades have brought major changes in the management of pain in the newborn....
Elemente actuale privind fiziopatologia cirozei hepatice la copil
Bogdan A. Stana, Evelina Moraru, Alice Azoicăi, Valentin Munteanu, Doina Mihăilă, Roxana Mihaela Barbu
Liver cirrhosis is a serious symptomatic complex, caused by progressive and generally irreversible liver damage, which causes the destruction of the lobular architecture by extensive fibrosis, nodular regeneration, and i...
Intoleranţa ereditară la fructoză: capcană de diagnostic la sugar?
Alina Grama, Claudia Sîrbe, Alexandra Mariş, Bogdan Bulata, Mariela Militaru, Tudor Lucian Pop
Hereditary fructose intolerance (HFI) – also called fructose-1-phosphate aldolase deficiency – is a rare inherited metabolic disorder characterized by the body’s inability to metabolize fructose or its precursors (sugar,...
Tusea cronică la copil – provocări
Mirela Pavelescu, Irina Dijmărescu, Daniela Păcurar
Introducere. Tusea reprezintă simptomul cel mai frecvent pentru consultaţia la medicul de familie în multe ţări din Europa. Persistenţa ei o perioadă îndelungată afectează calitatea vieţii pacientului. Prezentare de caz. Un adolescent de 13 ani este internat pentru tuse spastică frecventă, emetizantă, diur...