Arthrogryposis multiplex congenita (AMC) is a well-known newborn congenital anomaly, being characterized by multiple joint contractures. Amyoplasia is the most frequent form of AMC and has a markedly increased occurrence in one of the monozygotic twins. In AMC patients, besides contracture abnormalities seen especially in distal joints, other clinical features can be observed: micrognathia, low set years, short neck, broad nasal root and musculoskeletal system impairment. Arthrogryposis is defined by more than two affected joints, as clinical findings describe at birth. We present the case of a 34-week preterm male infant from a monochorionic monoamniotic twin pregnancy, with caesarean delivery, cephalic presentation, birth weight: 1580 g, with multiple spinal and limb malformations (the particular aspect of lower limbs with joint contractures and distal movement restriction of the upper limbs; scoliotic posture), decreased muscle tone and pneumoperitoneum.
Artrogripoza la un nou-născut prematur provenit din sarcină gemelară. Prezentare de caz
Arthrogryposis in a preterm newborn from twin pregnancy. Case report
First published: 07 mai 2018
Editorial Group: MEDICHUB MEDIA
DOI: 10.26416/Peri.2.1.2018.1648
Abstract
Rezumat
Artrogripoza congenitală multiplă (AMC) este caracterizată în literatura de specialitate de multiple contracturi articulare congenitale la nou-născut. Amioplazia este cea mai frecventă formă de AMC şi are o incidenţă crescută la unul dintre gemenii monozigoţi. La pacienţii cu AMC, pe lângă contracturile anormale observate la nivelul articulaţiilor distale, pot fi observate micrognaţia, urechile jos inserate, gâtul scurt, baza nasului lărgită şi diverse alte modificări ale sistemului musculoscheletal. Diagnosticul de artrogripoză se caracterizează prin fixarea a două sau mai multe articulaţii, obiectivat la naştere. Prezentăm cazul unui nou-născut prematur (34 de săptămâni), de sex masculin, provenit din sarcină gemelară, monocorială, monoamniotică, extras prin operaţie cezariană, prezentaţie craniană, greutatea la naştere: 1580 g, care prezintă malformaţii la nivelul coloanei vertebrale şi ale membrelor (postură particulară a membrelor inferioare, cu fixarea articulaţiilor şi limitarea mişcărilor, şi la nivelul membrelor superioare distal; postură particulară a trunchiului – aspect scoliotic), hipotonie musculară şi pneumoperitoneu.
Introducere
Artrogripoza congenitală multiplă (AMC) este caracterizată în literatura de specialitate de multiple contracturi articulare congenitale la nou-născut.
În 1841, Otto a descris prima dată această anomalie(1). În 1932, Seldon a publicat în Regatul Unit al Marii Britanii un articol în care a denumit-o „amioplazie congenitală”, subliniind dezvoltarea insuficientă a sistemului muscular(2). Termenul de artrogripoză este unul descriptiv, şi nu diagnostic.
Din punct de vedere genetic, Hall a descris cel puţin 150 de diagnostice posibile care pot duce la multiple deformări articulare la nou-născut(3).
AMC este o patologie congenitală caracterizată prin multiple contracturi articulare, cu o incidenţă de 1 din 3000 de naşteri(4).
În literatura de specialitate sunt descrise mai multe forme clinice. În forma clasică, denumită de Sheldon amioplazie congenitală, pot fi afectate toate cele patru membre sau doar cele inferioare.
Amioplazia este cea mai frecventă formă de AMC şi este mai frecvent întâlnită la unul dintre gemenii proveniţi din sarcină monocorială monoamniotică. O altă problemă legată de cazurile de AMC în sarcinile gemelare este diagnosticul complicaţiilor induse de sindromul transfuzor-transfuzat (TTS)(5-6).
Hall şi colaboratorii săi au descris o formă distală cu transmitere autozomal dominantă, în care atât mâinile, cât şi picioarele sunt grav deformate, cu contracturi minime, în timp ce coloana vertebrală prezintă riscul de a dezvolta scolioză(7). La naştere, copiii pot prezenta multiple deformări, iar părinţi sunt adesea îngroziţi de înfăţişarea propriului copil. În plus faţă de rigiditatea musculară şi contracturile articulare multiple, pielea este lipsită de cute şi pliuri peste articulaţiile caracteristice. Membrele sunt tubulare şi inexpresive, iar trunchiul poate fi afectat, dând aspectul caracteristic de „păpuşă din lemn”. De asemenea, pot apărea şi alte anomalii congenitale, cum ar fi criptorhidia, laparoschizisul sau atrezia intestinală(8).
În 1974, Pena şi Shokeir descriu un sindrom letal de anchiloze multiple, camptodactilie, anomalii faciale şi hipoplazie pulmonară la doi fraţi(9). Ulterior, sunt descrise tot mai multe cazuri sporadice şi familiale bine documentate cu fenotip similar(10). S-a observat o legătură între cazurile de AMC şi alte cazuri de trisomii, în special trisomia 18 (sindrom Edwards)(11-14). De aceea, în caz de necesitate, diagnosticul prenatal poate fi recomandat acestor pacienţi.
De cele mai multe ori, fenotipul rezultă din akinezie fetală, de aceea se impune obligatoriu o clasificare exactă a diferitelor tipuri întâlnite în practica medicală, în funcţie de semnele clinice. Astfel, se definesc termenii „fenotipul Pena-Shokeir” sau secvenţa „akinezie fetală”, întrucât aceştia nu definesc o singură trăsătură clinică, ci unul sau mai multe semne clinice(15). Limitarea mişcărilor fetale care se poate observa încă din primele săptămâni de gestaţie (în special în săptămânile 7 şi 8) este cel mai important factor etiologic(5). Principalele cauze de AMC cu implicarea limitării mişcărilor fetale incriminează factori care ţin de sistemul muscular (distrofie musculară, miastenia gravis la mamă), neurologici (anomalii ale creierului, măduvei spinării şi nervilor periferici) şi boli de ţesut conjunctiv (displazie distrofică, sindrom Marfan)(6,16).
De obicei, factorul etiologic al AMC poate fi evidenţiat la necropsie(17).
La pacienţii cu AMC, pe lângă contracturile anormale observate la nivelul articulaţiilor distale, pot fi observate şi alte anomalii, precum: micrognaţia, urechile jos inserate, gâtul scurt, baza nasului lărgită şi diverse modificări ale sistemului musculoscheletal(18,19).
Diagnosticul precoce şi înlăturarea factorilor externi care pot cauza AMC pot preveni deformările sau urgenta tratamentul acestora în perioada postnatală şi, astfel, grăbi recuperarea(18).
Diagnosticul clinic este facilitat de mişcările reduse sau absente ale fătului şi de postura persistent anormală a membrelor. Cu toate acestea, diagnosticul etiologic al acestor anomalii nu poate fi stabilit cu certitudine. Deşi componenta senzitivă nu este afectată, prognosticul afecţiunii este nefavorabil, prin multiplele deformări musculoscheletale şi comorbidităţi(5).
Prezentare de caz
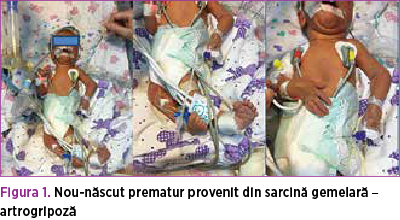
Prezentăm cazul unui nou-născut prematur (34 de săptămâni), de sex masculin, provenit din sarcină gemelară, monocorială, monoamniotică, extras prin operaţie cezariană, prezentaţie craniană, greutatea la naştere: 1580 g, scor Apgar 1 la 1 minut.
Menţionăm că nou-născutul se internează prin transfer într-un centru de chirurgie pediatrică dintr-o maternitate de nivel II la trei zile de viaţă pentru abdomen destins de volum, sensibil la palpare, asociind insuficienţă respiratorie, pentru care a necesitat suport ventilator invaziv.
Din istoric reţinem că, după manevrele de reanimare din sala de naşteri, se decide intubaţia şi ventilaţia mecanică. Se asigură confort termic şi se instituie tratament cu PEV de reechilibrare hidroelectrolitică şi antibioterapie. La aproximativ trei zile de viaţă se decide suprimarea suportului ventilator, însă la câteva ore prezintă un episod de desaturare severă şi este reintubat; în evoluţie asociază distensie marcată a abdomenului, iar pe sonda endotraheală se exteriorizează sânge proaspăt.
La internarea în TINN a secţiei de chirurgie pediatrică, nou-născutul prezintă stare generală extrem de gravă, este intubat şi ventilat mecanic, SpO2 = 88%, fluxul ventilatorului diminuat bilateral; de pe sonda endotraheală se aspiră sânge proaspăt în cantitate mare; AV = 130 b/min; abdomen destins de volum, hernie inghinală bilaterală; malformaţii la nivelul coloanei vertebrale şi al membrelor (postură particulară a membrelor inferioare, cu fixarea articulaţiilor şi limitarea mişcărilor în porţiunea distală a membrelor superioare; postură particulară a trunchiului – aspect scoliotic), hipotonie musculară (figura 1).
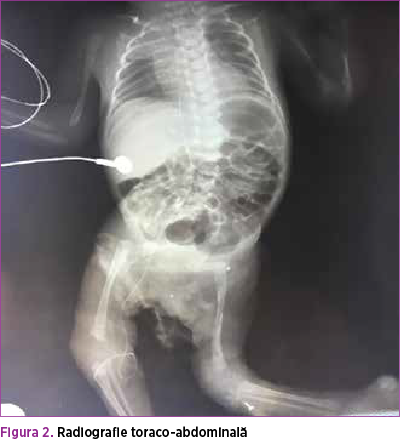
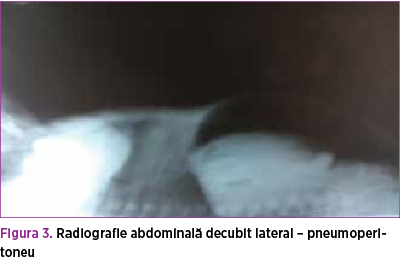
Radiografia toraco-abdominală evidenţiază atelectază în câmpul pulmonar drept şi hiperinflaţie în câmpul pulmonar stâng; pneumoperitoneu (figura 2 şi figura 3).
În TINN este ventilat mecanic, primeşte PEV de reechilibrare hidroelectrolitică şi antibioterapie, plasma proaspăt congelată (PPC). Medicul chirurg practică o laparotomie şi face irigaţii cu ser fiziologic, cu eliminare de meconiu în cantitate mică.
În evoluţie: se menţine intubat şi ventilat mecanic o lungă perioadă, continuă tratamentul cu PEV de rehidratare hidroelectrolitică, antibioterapie cu spectru larg şi antifungic profilactic, transfuzii multiple cu PPC şi ME. Suportul nutriţional este asigurat iniţial parenteral, ulterior enteral, fiind alimentat pe sonda nazogastrică cu formule de prematuri.
În perioada spitalizării în TINN s-a efectuat kinetoterapie zilnic. A fost extubat de câteva ori, însă de fiecare dată a prezentat secreţii salivare abundente, pe care nu le putea înghiţi. Unele dintre aceste episoade au fost urmate de obstrucţia căilor respiratorii şi soldate cu stop respirator, care a necesitat resuscitare şi reintubare orotraheală/nazotraheală. La vârsta de 4 luni şi o săptămână, intubat şi ventilat mecanic, starea generală se deteriorează brusc şi prezintă desaturări semnificative, urmate de bradicardie severă neresponsivă la manevrele de resuscitare cardiopulmonară.
Managementul acestui pacient a constat într-o abordare multidisciplinară, echipa fiind formată din medici neonatologi, pediatri, chirurgi, neurologi, ortopezi, medici de terapie intensivă şi kinetoterapeuţi.
În urma consultului de ortopedie pediatrică s-au stabilit următoarele diagnostice: artrogripoză, luxaţie congenitală de şold bilateral, picior strâmb varus equin bilateral, care s-au menţinut în evoluţie. Pentru aceste afecţiuni s-a efectuat kinetoterapie, cu reevaluare periodică şi temporizarea intervenţiei chirurgicale.
Consultul de neurologie pediatrică a pus în evidenţă: mişcări active prezente rar şi de mică amplitudine, limitate distal la nivelul membrelor superioare şi foarte limitate distal la nivelul membrelor inferioare, ROT abolite la toate nivelurile, hipotonie musculară, postură particulară a trunchiului – aspect scoliotic. Aceste modificări evidenţiate de examenul clinic neurologic sunt datorate patologiei ortopedice anterior menţionate. Din punct de vedere senzitiv-senzorial, nu se descriu anomalii. Prezintă grimase de plâns, reflex de vomă declanşat de aspirarea secreţiilor, deschide ochii spontan. În urma acestui consult se ridică suspiciunea de amiotrofie spinală, motiv pentru care se recomandă testare genetică. Rezultatul analizei genetice infirmă suspiciunea de amiotrofie spinală.
Întrucât pacientul face parte din categoria prematurilor, s-a efectuat consult oftalmologic, care nu a evidenţiat modificări patologice.
Discuţii
AMC este o patologie congenitală caracterizată prin contracturi articulare(5). Cu toate acestea, deşi contracturile articulare din AMC sunt tipic prezente la examinarea ecografică, ele sunt interpretate ca simple observaţii clinice; pe baza lor nu se poate pune un diagnostic de certitudine(12). Din acest motiv, 75% din cazurile de AMC rămân nediagnosticate. Astfel, ar fi utilă raportarea acestor cazuri de artrogripoză, pentru ca în viitor clinicienii să le acorde o atenţie deosebită la examinările ecografice şi la diagnostic(12). O altă utilitate a ecografiei prenatale o reprezintă identificarea modificărilor asociate cu AMC: imobilitate fetală, postură anormală, ventriculomegalie cerebrală, dismorfisme, restricţia de creştere intrauterină, creşterea translucenţei nucale, scolioza, pieptul mic, coastele subţiri(13).
Cazurile care provoacă limitarea spaţiului intrauterin sunt frecvent citate în etiologia AMC (sarcină gemelară, mioame, anomalii ale uterului). În cazul prezentat, dintre aceşti factori a fost luat în considerare mai ales particularitatea sarcinii, aveasta fiind gemelară, monocorială, monoamniotică, cauzând AMC prin restricţia spaţiului intrauterin.
Pe de altă parte, amioplazia este cea mai frecventă formă de AMC şi are o incidenţă crescută la unul din gemenii monozigoţi(20,21), fapt ce poate fi obiectivat şi în cazul prezentat.
Astfel, considerăm drept cea mai probabilă cauză a AMC în cazul evaluat ca fiind limitarea spaţiului intrauterin. Celelalte ipoteze etiologice (neurologică – amiotrofie spinală, genetică) nu au fost susţinute prin evaluările efectuate. Din acest motiv, prognosticul cognitiv şi senzitivo-senzorial a fost favorabil (elemente evidenţiate şi în evaluarea neurologică); în schimb, prognosticul rezervat era marcat de: prematuritate, complicaţiile asociate imobilizării prelungite, detresă respiratorie marcată care a necesitat ventilaţie mecanică prelungită, asocierea de multiple scheme de antibioterapie cu spectru larg, utilizarea cateterelor venoase centrale menţinute un timp îndelungat. Toţi aceşti factori au contribuit la creşterea riscului infecţios. În ciuda eforturilor echipei multidisciplinare, s-a produs decesul pacientului.
În concluzie, AMC este o boală multifactorială cu morbiditate şi mortalitate fetală ridicate şi impact economic marcat în perioada pre- şi postnatală.
O varietate de sfaturi ar trebui să fie date părinţilor cu sarcini complicate cu AMC, incluzând istoria naturală a bolii, precum şi opţiunile de management, riscurile şi beneficiile. Datorită similarităţii cazurilor de AMC, observate mai ales în sarcinile gemelare cu TTS, pacienţii ar trebui evaluaţi atent şi simptomatologia TTS cunoscută.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
2. Sheldon W. Amyoplasia congenita (multiple congenital articular rigidity: arthrogryposis multiplex congenita). Arch Dis Child. 1932; 7:117–136.
3. Hall JG. Genetic aspects of arthrogryposis multiplex congenita. Clin Orthop Rel Res. 1985;194:44–53.
4. Hoff JM, Daltveit AK, Gilhus NE. Artrogryposis multiplex congenita - a rare fetal condition caused by maternal myasthenia gravis. Acta NeurolScand Suppl. 2006;183: 26-27.
5. Lowry RB, Sibbald B, Bedard T, Hall JG. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res A Clin Mol Teratol. 2010;88: 1057-1061.
6. Hall JG. Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach and general aspects. J Pediatr Orthop B. 1997;6: 159-166.
7. Hall JG, Reed SD, Green G. The distal arthrogryposis; delineation of new entries: review and nosologic discussion. Am J Med Gen. 1982;11:185–239.
8. Bevan WP, Hall JG, Baushad M, Strakeli LJ, et al. Arthrogryposis multiplex congenita (amyoplasia) (An orthopaedic perspective). J Pediatr Orthop. 2007;27:594–598.
9. Pena SD and Shokeir MH. Syndrome of camptodactyly, multiple ankyloses, facial anomalies, and pulmonary hypoplasia: a lethal condition. J Pediatr. 1974;85(3): p. 373-5.
10. Hall JG. Pena-Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Res A Clin Mol Teratol. 2009;85(8): p. 677-94.
11. Hoff JM, Loane M, Gilhus NE, Rasmussen S, Daltveit AK. Arthrogryposis multiplexa congenita: an epidemiologic study of nearly 9 million births in 24EUROCAT registers. Eur J Obstet Gynecol Reprod Biol. 2011;159: 347-350.
12. Filges I, Hall JG. Failure to identify antenatal multiple congenital contractures and fetal akinesia - proposal of guidelines to improve diagnosis. Prenat Diagn. 2013;33: 61-74.
13. Kalampokas E, Kalampokas T, Sofoudis C, Deligeoroglou E, Botsis D. Diagnosing arthrogryposis multiplex congenita: a review. ISRN Obstet Gynecol. 2012;264918.
14. Naguib KK, Al-Awadi SA, Moussa MA, Bastaki L, Gouda S, et al. Trisomy 18 in Kuwait. Int J Epidemiol. 1999;28: 711-716.
15. Hall JG. Analysis of Pena Shokeir phenotype. Am J Med Genet. 1986;25(1): p. 99-117.
16. Bonilla-Musoles F, Machado LE, Osborne NG. Multiple congenital contractures (congenital multiple arthrogryposis). J Perinat Med. 2002;30: 99-104.
17. Hoff JM, Loane M, Gilhus NE, Rasmussen S, Daltveit AK. Arthrogryposis multiplexa congenita: an epidemiologic study of nearly 9 million births in 24 EUROCAT registers. Eur J Obstet Gynecol Reprod Biol. 2011;159: 347-350.
18. Banker BQ. Arthrogryposis multiplex congenita: spectrum of pathologic changes. Hum Pathol. 1986;17: 656-672.
19. Hall JG. Overview of arthrogryposis. In: Staheli LT, Hall JG, Jaffe KM, Pahoike DE, eds. Arthrogryposis: A Text Atlas. Cambridge: Cambridge University Press. 1998;1–25.
20. Hall JG, Reed SD, McGillivray BC, Herrmann J, Partington MW, et al. Part II. Amyoplasia: twinning in amyoplasia - a specific type of arthrogryposis with an apparent excess of discordantly affected identical twins. Am J Med Genet. 1983;15: 591-599.
21. Hall JG, Aldinger KA, Tanaka KI. Amyoplasia revisited. Am J Med Genet A. 2014;164A: 700-730.
Articole din ediţiile anterioare
Bloc atrioventricular congenital la nou-născut din mamă cu lupus eritematos sistemic - prezentare de caz
Introducere. Blocul atrioventricular congenital (BAVC), în absenţa unor malformaţii sau anomalii cardiace structurale majore, este datorat cel mai ...
Semnificaţia gazelor din cordonul ombilical în encefalopatia nou-născutului – review al literaturii
Studii experimentale şi modele animale au arătat că encefalopatia hipoxic-ischemică este un proces în evoluţie, mai degrabă, decât un eveniment sin...
Sindromul Prader-Willi - prezentare de caz şi revizuirea literaturii
Sindromul Prader-Willi este consecinţa mai multor defecte genetice în regiunea 15q11-q13, între care şi modificările de metilare, şi este asociat l...
Managementul durerii la nou-născut
Progresele efectuate în ultimii ani au stabilit că nou-născutul poate experimenta durere acută sau cronică, iar managementul corect are beneficii a...