Sindromul hiper-IgE − între provocări clinice şi complexitate patogenică
Hyper IgE syndrome − between clinical challenges and pathogenic complexity
Abstract
Hyper-IgE syndrome is a complex primary immunodeficiency involving the immune system, bones, connective tissue and teeth. Along with the high levels of total IgE, syndromic complexity is due to mutations in the STAT3 gene, with subsequent changes in cytokine cascades, with resonance in immunological and somatic changes. The authors describe the essential features of hiper IgE syndrome, exemplified by a clinical case.
Keywords
hyper-IgEchildallergyRezumat
Sindromul hiper-IgE este o imunodeficienţă primară complexă, ce implică sistemul imun, oasele, ţesutul conjunctiv şi dinţii. Alături de valorile mult crescute ale IgE totale, complexitatea sindromatică este dată de mutaţii în gena STAT3, cu modificări ulterioare asupra cascadelor citokinice, având răsunet asupra modificărilor imunologice şi somatice. Autorii descriu trăsăturile esenţiale ale sindromului hiper-IgE, exemplificate printr-un caz clinic prezentat.
Cuvinte Cheie
hiper-IgEcopilalergieSindromul hiper-IgE este o imunodeficienţă primară complexă, caracterizată printr-un spectru de anomalii care interesează sistemul imun, oasele, ţesutul conjunctiv şi dinţii. Este caracteristică triada clinică:
-
niveluri serice crescute de IgE (>2000 UI/ml);
-
abcese stafilococice recurente ale pielii;
-
pneumonie caracterizată prin complicaţia cu pneumatocel.
Sindromul hiperimunoglobulinei E (HIES) autozomal-dominant a fost descris pentru prima dată ca sindromul Iov, în 1966(1), şi a inclus triada eozinofilie, eczeme şi infecţii recurente ale pielii şi pulmonare (denumit după personajul biblic Iov).
Nivelul crescut al IgE a fost recunoscut ca o caracteristică esenţială a sindromului în 1972, iar numele HIES a fost propus ulterior(2). Fenotipul a fost extins pentru a include multe ţesuturi conjunctive şi anomalii scheletice.
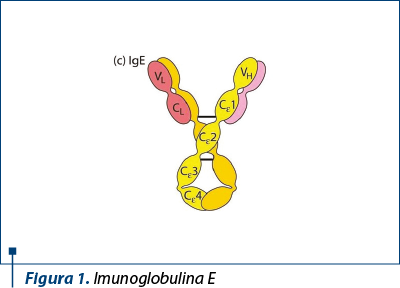
Imunoglobulinele E se găsesc în principal în secreţiile mucoase de la nivelul tractului gastrointestinal şi respirator, în ser fiind prezente în concentraţii foarte mici.
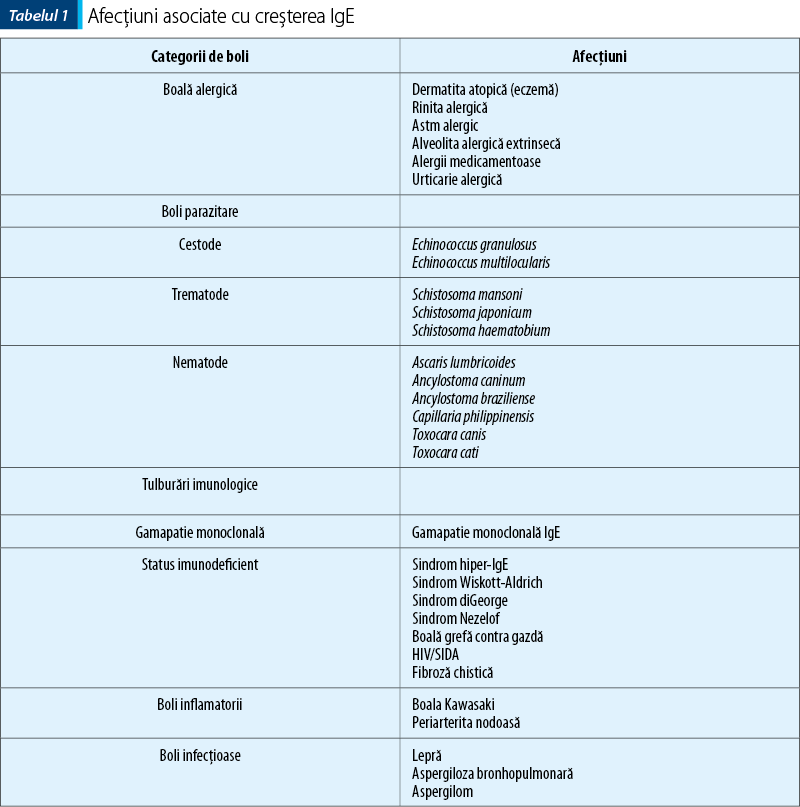
Creşteri ale nivelului IgE sunt înregistrate în afecţiuni alergice, boli parazitare, boala Hodgkin şi mielom multiplu de tip IgE.
Manifestările clinice majore ale afecţiunilor alergice IgE mediate sunt anafilaxia, astmul, rinita alergică, dermatita atopică, alergia alimentară, alergia la înţepături de insecte şi alergia la lapte.
Mai recent, au fost descrise anomalii vasculare, inclusiv anevrism coronarian fără ateroscleroză, şi anomalii ale RMN-ului cerebral, inclusiv hiperintensiunile focale şi malformaţiile Chiari I.
În 2007, mutaţiile dominante autozomale în traductorul de semnal şi activatorul genei transcripţie-3 (STAT3) au fost identificate ca fiind cauza moleculară a acestei boli(4,5,6).
Fiziopatologie
Mutaţiile în gena STAT3 au fost identificate în aproape toate cazurile de HIES verificate clinic(7,8). STAT3 este una dintre cele 7 proteine STAT (transductor de semnal şi activator de transcriere), care sunt mesageri critici secundari pentru mulţi receptori de citokine, hormoni şi factori de creştere. În general, tirozin-kinazele familiei Janus (JAKs) se leagă la componentele intracelulare ale receptorilor citokinici şi sunt, la rândul lor, legate de STAT la semnalizarea citokinelor. Atunci când citokinele se leagă de receptorul lor conjugat, JAKs fosforilează receptorul citokinic şi ulterior STAT, care apoi disociază de complexul JAK-receptor. STAT-urile fosforilate se dimerizează în citozol prin domeniile fosfotirozinelor şi domeniile de omologie Sr2 (SH2). STAT-urile dimerizate se translocă apoi în nucleu, unde se leagă de ADN în secvenţele promotor ale genelor-ţintă pentru a activa transcripţia. Această activitate este, de obicei, oprită prin defosforilare STAT. STAT3 este localizat pe cromozomul uman 17q21.
Mutaţiile STAT3 au fost găsite la persoane de origine asiatică, africană, caucaziană şi hispanică şi se corelează bine cu boala HIES. Mutaţiile sunt predominant inversii şi deleţii care duc la producerea proteinelor cu activitate negativă dominantă. Nu au fost identificate până în prezent alele, ceea ce indică faptul că haploinsuficienţa nu este un mecanism de cauzalitate a bolii.
Majoritatea mutaţiilor HIES STAT3 sunt localizate în regiunile de legare a SH2 şi ADN. Mutantele negative dominante antagonizează proteina de tip sălbatic, conducând la o activitate STAT3 mai mică de 50%.
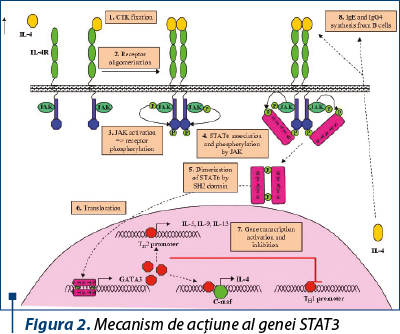
Prin urmare, pacienţii cu HIES au activitate STAT3 mai mică de 50%, ceea ce este suficient pentru a susţine viaţa şi multe aspecte ale dezvoltării, dar neadecvat pentru funcţia imună normală sau remodelarea normală a ţesuturilor.
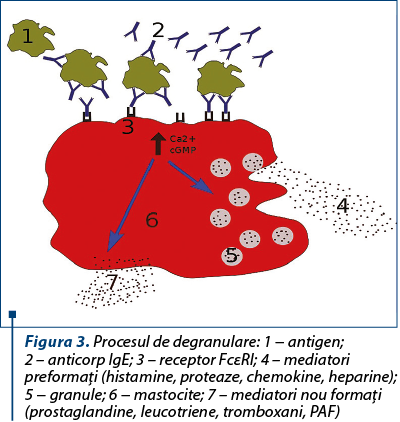
Din cauza afectării semnalizării IL-6 şi IL-23 prin STAT3, factorul crucial de transcripţie a receptorului (ROR-gt) este diminuat, afectând expresia IL-17 şi diferenţierea Th17(9). Celulele Th17 sunt celule T helper, care produc IL-17, o citokină importantă în recrutarea şi activarea neutrofilelor şi apărarea împotriva fungilor şi a bacteriilor extracelulare. Celulele Th17 produc, de asemenea, IL-22, care stimulează producţia epitelială a defensinelor b, peptide mici, cruciale pentru uciderea bacteriilor şi a ciupercilor. Aceste citokine, care sunt inadecvat crescute în HIES, pot explica o parte din sensibilitatea la infecţii a pielii şi epiteliului pulmonar.
Inflamaţia excesivă în HIES este sugerată de dezvoltarea frecventă a pneumatocelului după pneumonii, în ciuda terapiei adecvate a agenţilor patogeni. Studiile de transcripţie şi studiile de producere a citokinelor au evidenţiat creşterea citokinelor proinflamatorii, cum ar fi factorul de necroză tumorală alfa (TNFa), interferonul gamma (IFNg) şi IL-12 în celulele HIES comparativ cu cele normale.
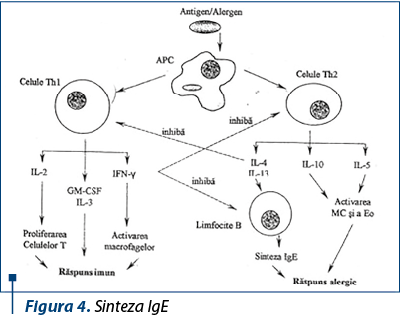
Nivelul anormal de scăzut al IL-10, o citokină critică pentru atenuarea răspunsului inflamator, poate, de asemenea, să contribuie la răspunsul inflamator exagerat în HIES. Semnalarea IL-10 este mediată prin STAT3. IL-10 are o reglare negativă a feedbackului macrofagelor, care exprimă costimulatori ce amplifică activarea celulelor T şi secretă citokine precum IL-12 şi IL-23 pentru a îmbunătăţi imunitatea mediată de celulă. Prin urmare, producţia şi semnalarea IL-10 afectată poate fi asociată cu creşterea citokinelor proinflamatorii. Poate chiar contribui la creşterea IgE şi a eozinofiliei la HIES din cauza lipsei supresiei normale a IL-4.
Sindromul hiper-IgE prezintă trei forme:
I. Forma autozomal-dominantă
II. Forma autozomal-recesivă − rară
III. Forma sporadică.
Epidemiologie
Frecvenţă
Statele Unite ale Americii
HIES este o boală rară, deşi incidenţa exactă este necunoscută. Nu are nicio asociere cunoscută cu rasa, etnia sau sexul.
Mortalitatea/morbiditatea
HIES este asociat cu o durată de viaţă variabilă, cu decese care au loc preponderent la maturitate, din cauza consecinţelor infecţiilor cronice. Într-o revizuire recentă a unei cohorte mari de pacienţi, vârsta medie a pacientului viu a fost de 27 de ani, dar vârstele au variat de la 3 la 58 de ani. Infecţiile legate de deces au apărut la o vârstă medie de 29 de ani.
Decesul la pacienţii cu HIE se datorează cel mai frecvent infecţiilor pulmonare cronice, care duc la apariţia pneumatocelului sau a bronşiectaziei sau la ambele. Unele specii de Aspergillus sau Scedosporium pot invada vasele de sânge pulmonare, ducând la răspândirea metastatică sau la hemoptizie fatală. Aceste infecţii sunt oportunişti secundari în regiunile pulmonare distruse, cauzate de vindecarea necorespunzătoare a infecţiilor anterioare(10).
Pacienţii cu HIES prezintă un risc crescut pentru limfom Hodgkin sau non-Hodgkin(11).
Rasă
Mutaţiile STAT3 au fost detectate la persoanele de origine caucaziană, africană, asiatică şi hispanică. Deşi cele mai multe cazuri documentate sunt la pacienţii de rasă albă, acest lucru se datorează, probabil, loturilor de populaţie studiată.
Sex
Nu au fost descrise diferenţe de gen în ceea ce priveşte severitatea sau incidenţa bolii.
Vârstă
Vârsta medie a diagnosticului a fost de 11,5 ani. Cu toate acestea, diagnosticul se poate face în copilărie sau la vârsta adultă. Supravieţuirea a fost ameliorată de-a lungul anilor, împreună cu o gestionare îmbunătăţită, permiţând pacienţilor să aibă copii. Având în vedere natura dominant-autozomală a HIES, aceşti descendenţi ai pacienţilor cunoscuţi primesc diagnosticul precoce şi terapia.
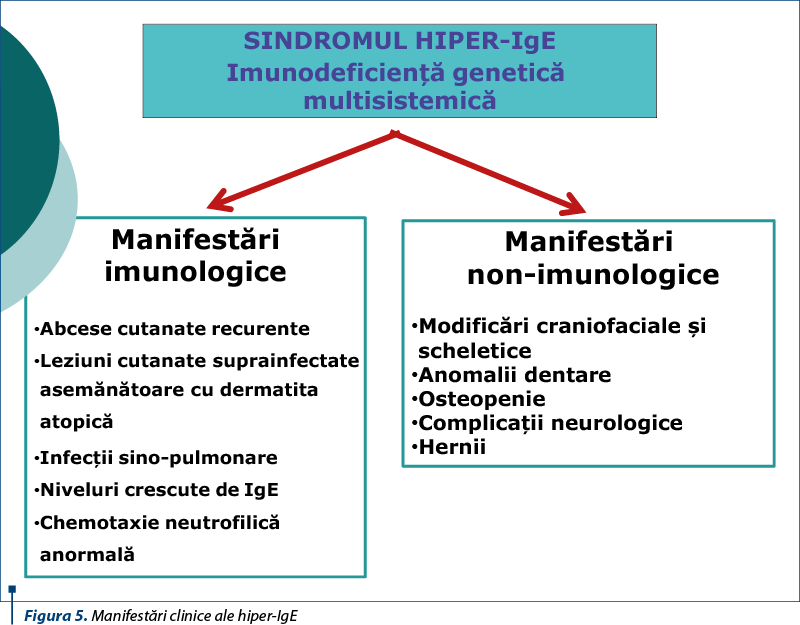
Istoric
Dermatita sub formă de eczemă începe în perioada de nou-născut şi este asociată şi condusă de infecţia cu Staphylococcus aureus. Abcesele cutanate datorate S. aureus sunt frecvente şi de obicei nu sunt asociate cu un răspuns inflamator important local sau sistemic, prin urmare pentru acestea se utilizează termenul de abces rece. Candidoza mucocutanată, inclusiv onicomicoza, apare frecvent. În schimb, verucile severe şi speciile de Molluscum nu sunt trăsături ale formei autozomal-dominante (AD) a HIES.
Pneumoniile încep din copilărie, de obicei din cauza S. aureus, Streptococcus pneumoniae şi Haemophilus influenzae. Similar cu abcesele pielii, semnele de toxicitate sistemică sunt modeste. Chiar şi atunci când sunt tratate agresiv şi prompt, pneumoniile se remit adesea, cu formarea pneumatocelului, probabil din cauza inflamaţiei locale necorespunzatoare şi a distrugerii ţesuturilor rezultate. Ulterior, superinfecţia pneumatocelelor cu Pseudomonas aeruginosa şi a mucegaiurilor, precum speciile Aspergillus şi Scedosporium, poate fi asociată cu morbiditate şi mortalitate semnificativă.
Infecţii oportuniste, inclusiv cu Pneumocystis jiroveci, histoplasmoză diseminată, criptococoză şi coccidioidomicoză, apar, dar sunt mai puţin frecvente. Sinuzita bacteriană recurentă şi otita sunt frecvente, dar suspiciunea de agenţi patogeni virali nu este crescută.
HIES este, de asemenea, asociat cu anomalii scheletice şi de ţesut conjunctiv, incluzând scolioza, hiperextensibilitatea, fracturile patologice, dentiţia primară reţinută, craniosinostoza şi anomaliile vasculare.
Mulţi pacienţi au hiperextensibilitatea cel puţin a unei articulaţii. În ciuda acestor anomalii ale ţesutului conjunctiv, pacienţii HIES prezintă, de obicei, o vindecare normală a rănilor. Majoritatea pacienţilor prezintă cel puţin o fractură netraumatică, cum ar fi coaste rupte după tuse. Osteoporoza este frecventă, dar nu pare să se coreleze cu frecvenţa fracturilor. Retenţia dinţilor primari se datorează eşecului exfoliaţiei primare a dinţilor(12); bolta ogivală, modificări ale mucoaselor orale şi limbii sunt adesea prezente. Tortuozitatea arterială şi anevrismul apar, de regulă, pe vasele mijlocii, cum ar fi arterele coronare(13). Anomaliile RMN ale creierului includ hiperintensiunile focale şi o incidenţă crescută a malformaţiilor Chiari I(14).
În ciuda creşterii IgE şi a eozinofiliei, pacienţii nu au, în general, atopie semnificativă în ceea ce priveşte alimentele sau alergenii sezonieri.
Un istoric detaliat al familiei poate ajuta la identificarea altor cazuri, dar majoritatea cazurilor sunt mutaţii spontane sau dominant-autozomale. Diagnosticul clinic al HIES s-a bazat pe un profil compozit al caracteristicilor imunologice şi non-imunologice, conducând la un scor care se corelează bine cu identificarea mutaţiilor STAT3.
Manifestări clinice
Pacienţii au un aspect facial caracteristic, cu un nas larg, cu ochii adânci, cu frunte lată. Este adesea prezent un palat înalt arcuit, iar dentiţia dublă poate fi prezentă. Membrana timpanică poate fi cicatricială din otita recurentă.
Atât pacienţii pediatrici, cât şi cei adulţi pot prezenta o erupţie cutanată eritematoasă, scuamoasă, tipică de dermatită eczematoasă. Sunt prezente cicatrice de la incizie şi drenaj al abceselor reci. Unghiile, vaginul şi regiunea inghinală sunt adesea afectate de candidoză.
Examenul pulmonar variază în funcţie de infecţiile pulmonare anterioare şi de modificările parenchimului pulmonar. Examenul variază de la murmur fiziologic normal până la dispariţia acestuia şi reducerea schimbului de aer asociat cu pneumatocelele mari. Curbarea unghiilor este obişnuită.
Scolioza >10 grade se observă la 60% dintre pacienţi. Hiperextensibilitatea a cel puţin unei articulaţii este prezentă la 70% dintre pacienţi.
Cauze
Majoritatea (dacă nu toate) cazurile de HIES dominant-autozomal se datorează mutaţiilor în STAT3, o moleculă-cheie de semnalizare pentru transcripţia genelor imunologice şi somatice.
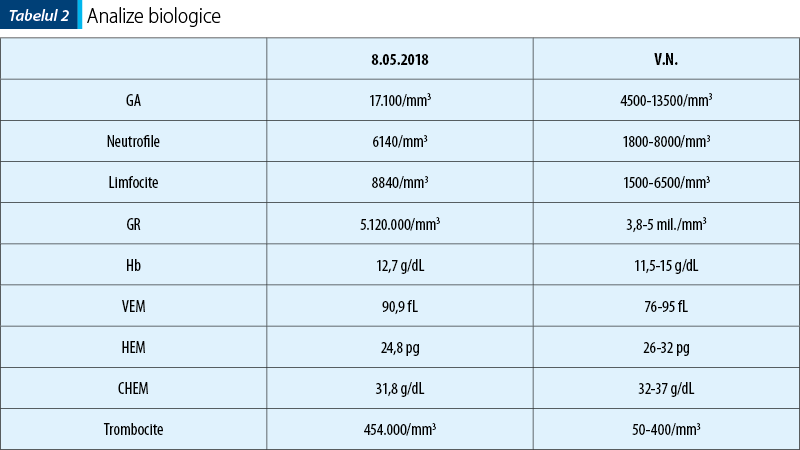
Examene de laborator
Hemoleucograma poate evidenţia eozinofilie. Eozinofilia însoţeşte, de obicei, creşterea IgE, dar nu este corelată cu aceasta. Numărul limfocitelor absolute este, de obicei, normal. Anemia şi trombocitopenia nu sunt în mod obişnuit asociate cu sindromul hiperimunoglobulinei E (HIES).
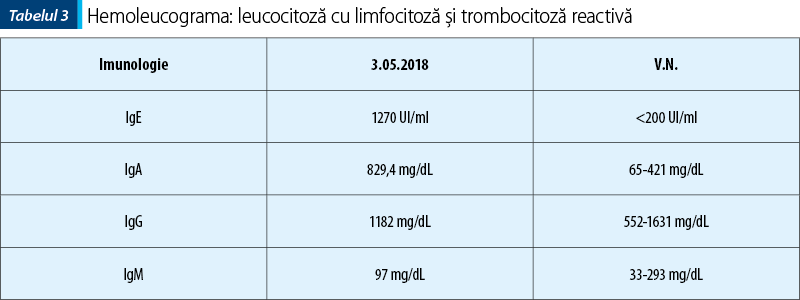
Imunoglobulinele serice IgG, IgA, IgM, IgE: IgE seric este de obicei >2000 UI/ml, în timp ce alte niveluri de imunoglobulină sunt normale. Totuşi, nivelul IgE se normalizează la vârsta adultă în aproximativ 20% din cazuri.
La copii, creşteri ale IgE peste 20 U/mL susţin diagnosticul de manifestare alergică; totuşi, o valoare normală nu exclude o boală alergică.
La adulţi, IgE sunt mai puţin folositoare în stabilirea etiologiei alergice a simptomelor:
-
IgE>1 DS peste medie sugerează boală alergică
-
IgE>2 DS peste medie susţin puternic etiologia alergică.
Cuantificarea nivelului IgE nu primează dacă etiologia alergică a fost probată clinic, iar niveluri crescute ale IgE au valoare limitată în predicţia tendinţei alergice. Creşterea IgE se regăseşte în multe patologii inflamatorii şi infecţioase.
Se poate vorbi despre sindrom hiper-IgE în următoarele situaţii:
-
Adulţi: >2000 UI/ml – definitorii, în prezenţa fenomenelor inflamatorii sau a pneumoniei.
-
La copii, care au niveluri în mod normal scăzute, sindromul hiper-IgE se consideră la valori de 10 ori mai crescute ale IgE pentru vârstă.
-
Cordon ombilical: <1 U/mL (1 U=2,4 ng).
Nivelurile stabile ca la adult se ating la vârsta de 5-7 ani; între 10 şi 14 ani se poate depăşi nivelul de la adult, iar după 70 de ani scad uşor.
Imunoglobulinele calitative pentru antigene specifice pot fi utile la pacienţii cu boală sino-pulmonară severă şi la cei care dezvoltă infecţii recurente, în pofida utilizării eficiente a profilaxiei antimicrobiene.
Unii pacienţi cu HIES au un răspuns scăzut de anticorpi specifici la vaccinurile pentru Haemophilus influenzae tip b (HiB), Streptococcus pneumoniae, tetanos sau difterie.
VSH-ul şi proteina C reactivă (CRP) sunt utile dacă nivelurile sunt ridicate, dar nivelurile normale nu exclud infecţia bacteriană.
Analizele privind electroliţii, coagularea şi funcţiile renală şi hepatică nu sunt afectate în HIES.
Abcesele cutanate trebuie incizate şi drenate şi se vor obţine culturi din secreţia de drenaj.
Atunci când infecţia pulmonară este prezentă, sputa trebuie obţinută pentru examinare.
Analiza mutaţiei STAT3 ar trebui efectuată pentru a confirma o suspiciune clinică ridicată a HIES.
Imagistică
Radiografia toracică, CT-ul sau ambele sunt necesare pentru a determina gradul de implicare a parenchimului pulmonar(16) (figura 6).
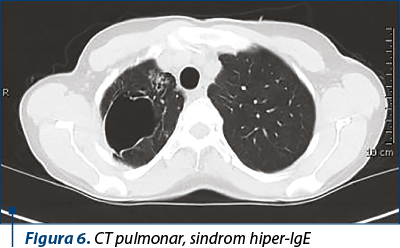
Pneumonii recurente, în special cele datorate S. aureus, pot conduce la formarea pneumatocelelor. Pneumatocelele, cum ar fi cele demonstrate în figura 6, pot permite apoi superinfecţia fungică.
Trebuie efectuate radiografii ale coloanei vertebrale pentru a evalua scolioza. De asemenea, trebuie efectuate radiografii osoase sau articulare din cauza riscului crescut de fracturi patologice la aceşti pacienţi.
Anomaliile vasculare sunt, de obicei, asimptomatice, incluzând anevrisme intracraniene şi ectazia arterei coronare, tortuozitatea şi anevrismele. Aceste anomalii sunt frecvente, detectate de CT şi RMN în studiile de cercetare. Studiile cu raze X sau IRM ale craniului arată adesea craniosinostoză.
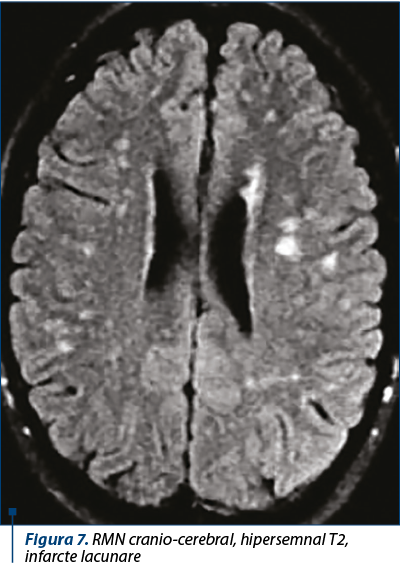
RMN-ul cerebral prezintă, de obicei, hipersemnal T2, o prevalenţă crescută a infarctelor lacunare şi o rată crescută de malformaţii Chiari tip I.
Hipertensiunea intracraniană a fost observată cu frecvenţă crescută la pacienţii de toate vârstele cu sindrom Iov.
Diagnostic diferenţial
Diagnosticul diferenţial al creşterii IgE se realizează pe baza scorului de severitate al National Institute of Health (NIH).
-
Parametri clinici
-
Abcese cutanate
-
Pneumonii
-
Anomalii de parenchim pulmonar (pneumatocele, bronşiectazii)
-
Infecţii grave
-
Rash (nou-născut)
-
Eczeme
-
Sinuzite/otite
-
Candidoze
-
Dentiţie dublă
-
Scolioză
-
Fracturi spontane
-
Facies caracteristic
-
Bolta palatină ogivală
-
Distanţa intermolară crescută
-
Hiperlaxitate reticulară
-
Anomalii congenitale
-
Limfom
-
Osteopenie
-
Număr anormal de coaste
-
-
Parametri biologici
-
IgE crescute
-
Eozinofilie
-
-
Scor
-
>40: sugestiv pentru sindrom hiper-IgE – severitate crescută
-
20-40: severitate medie
-
<20: puţin probabil sindrom hiper-IgE.
-
Tratament
Îngrijirea pacienţilor cu sindrom de hiperimunoglobulină E este îndreptată în principal spre tratarea şi prevenirea infecţiilor recurente ale pielii şi pulmonare. Antimicrobienele profilactice, cum ar fi trimetoprim/sulfametoxazol, sunt foarte eficiente. Cu prevalenţa crescândă a S. aureus rezistent la antibiotice şi a altor agenţi patogeni, este important ca sensibilităţile culturii microbiene să fie obţinute în mod activ pentru terapiile antimicrobiene directe. Deoarece pacienţii au adesea semne minime de toxicitate sistemică, pragul pentru examinarea infecţiei active care necesită tratament agresiv ar trebui să fie redus.
Controlul dermatitei este esenţial pentru scăderea frecvenţei infecţiilor cutanate şi îmbunătăţirea calităţii vieţii, în special pentru pacienţii pediatrici cu dermatită mai severă. Expunerea la concentraţii scăzute de clor, cum ar fi cea obţinută prin înotul frecvent într-o piscină clorinată sau prin băile de albire (jumătate de ceaşcă de înălbire într-o cadă de apă timp de 15 minute, de trei ori pe săptămână), este foarte eficientă pentru controlul infecţiilor cutanate stafilococice.
Candidoza mucocutanată şi onicomicoza sunt tratate după necesităţi cu terapie antifungică, cel mai frecvent fluconazol.
Extinderea acoperirii antifungice pentru a include specia Aspergillus (itraconazol, voriconazol, posaconazol) trebuie luată în considerare pentru persoanele cu pneumatocel.
Vaccinul antigripal anual este recomandat, deşi unii autori dezaprobă administrarea vaccinurilor cu agenţi vii atenuaţi.
În prezent, nu există recomandări clare pentru terapia medicală de rutină a anomaliilor vasculare, musculo-scheletice şi ale ţesutului conjunctiv asociate cu tulburarea. Transplantul de măduvă osoasă sau de celule stem a fost utilizat în prea puţine cazuri pentru a fi sigur de siguranţa sau valoarea sa în HIES.
Caz clinic:
O.C.V., de sex masculin, în vârstă de 10 ani, provenit din mediul rural, este internat la 7.05.2018 pentru tuse productivă.
Dintre antecedentele familiale amintim: mama − 33 de ani, tatăl − 39 de ani, aparent sănătoşi, neagă contactul cu boli infecto-contagioase sau TBC. Antecedente fiziologice: rang 1, născut natural, la termen, Gn=2900 g; APGAR 9; nu a prezentat icter fiziologic, alimentat natural; vaccinat în maternitate şi la vârsta de două luni.
Antecedente medicale patologice:
-
Meningită bacteriană+fungică la vârsta de trei luni, care s-a soldat cu sechele importante: encefalopatie cronică infantilă, hidrocefalie severă triventriculară, retard neuro-psihomotor grav, epilepsie generalizată grandmall.
-
În 2015 (la vârsta de 8 ani) se diagnostichează un chist aerian pulmonar necomplicat, cu ocazia unui episod infecţios pulmonar.
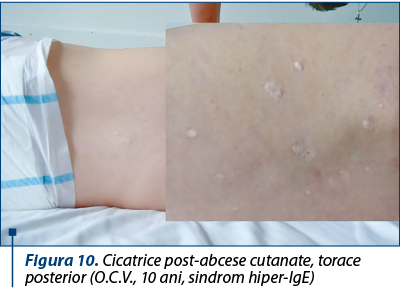
Figura 10. Cicatrice post-abcese cutanate, torace posterior (O.C.V., 10 ani, sindrom hiper-IgE) -
Stafilococie cutanată (abcese multiple), soldată cu cicatrice post-vindecare.
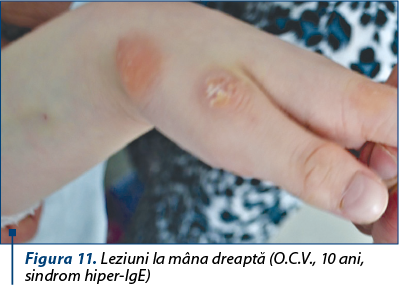
Figura 11. Leziuni la mâna dreaptă (O.C.V., 10 ani, sindrom hiper-IgE) -
Sindrom hiper-IgE, asociat cu multiple episoade infecţioase respiratorii.
Istoricul bolii: ne aflăm în faţa unui copil în vârstă de 10 ani, imobilizat la pat, cunoscut cu encefalopatie cronică infantilă şi retard neuro-psihomotor sever, în tratament cronic cu levitiracetam 4 ml x 2/zi, Depakine® 5 ml x 2/zi, Rispolept® 0,5 ml/zi şi Rivotril® 1 cp x 2/zi.
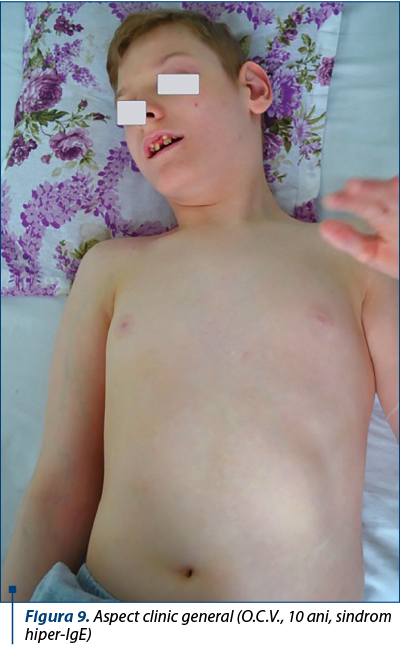
Examen clinic – la internare, observăm o stare generală influenţată, copilul având talie şi greutate în limite normale vârstei (T=132 cm, G=36 kg; IMC=20,7 kg/mp), cu o stare de nutriţie bună, un facies uşor asimetric; tegumente palid-elastice; mucoase normal colorate; multiple cicatrice post-abcese cutanate pe toracele posterior.
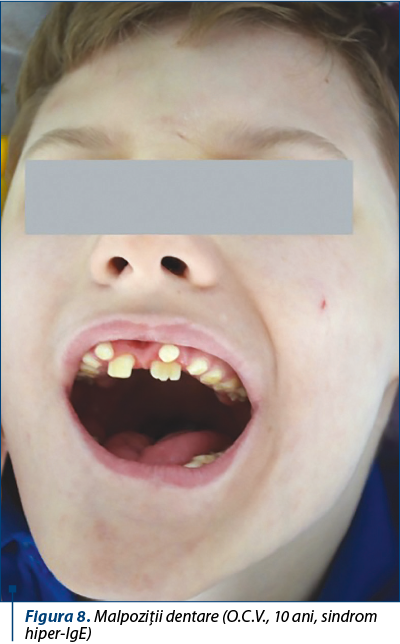
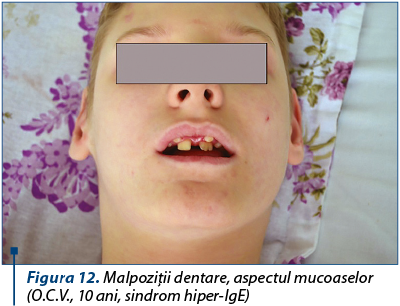
La nivelul cavităţii orale se observă malpoziţii dentare cu dentiţie dublă.
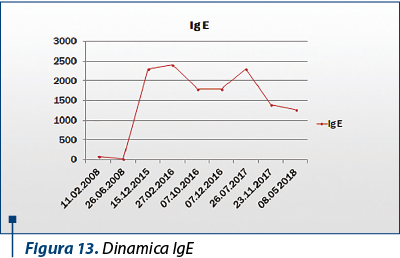
IgE în dinamică au arătat valori de până la 2500 UI/ml, menţinându-se la un plafon ridicat, de aproximativ 15 ori limita superioară a normalului la reevaluări repetate.
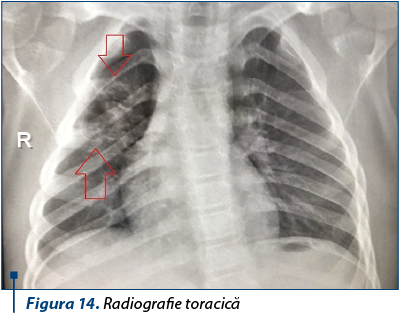
Radiografia toracică a arătat o formaţiune radio-transparentă bine delimitată, cu perete subţire, cu dimensiuni de 5,5/3,7 cm, localizată la nivelul lobului superior drept, interpretată drept chist aerian necomplicat.
-
CT toracic (7 decembrie 2016)
-
formaţiune chistică cu perete subţire şi conţinut strict aeric, polilobată, cu diametrele de 27/19/31 mm, situată în LSD.
-
-
CT cranio-cerebral (7 decembrie 2016)
-
hidrocefalie severă triventriculară simetrică
-
hipoplazie importantă de corp calos
-
sinuzită maxilară stângă.
-
-
Diagnostic pozitiv
-
Encefalopatie cronică infantilă
-
Retard neuro-psihomotor sever
-
Epilepsie generalizată
-
Traheobronşită acută
-
Chist aerian pulmonar necomplicat
-
Malpoziţie dentară – dentiţie dublă
-
Sindrom hiper-IgE
-
Spasmofilie.
-
-
Diagnostic diferenţial:
-
sindrom Wiskott Aldrich – exclus prin aspectul sever al dermatitei, numărul şi aspectul normal al trombocitelor, absenţa sindromului hemoragipar;
-
boală granulomatoasă cronică – boală X-linkată, care se prezintă cu infecţii severe cutanate, osoase, viscerale, dar nu asociază dermatită, valori crescute ale IgE totale; diagnosticul de certitudine este susţinut de valoarea patologică a testului NBT;
-
sindrom Omenn, caracterizat prin imunodeficienţă severă combinată, ce asociază eritrodermie severă, diaree, hepatosplenomegalie, hipereozinofilie şi creştere marcată a IgE, dar cu valori extrem de scăzute sau absente ale celorlalte clase de Ig serice;
-
imunodeficienţă comună variabilă – valori normale ale IgE, dermatită mai puţin severă;
-
dermatită atopică – exclusă prin lipsa altor alergii.
-
Tratament
Nu există tratament specific pentru sindromul hiper-IgE în prezent.
Tratamentul se bazează pe administrarea unei medicaţii antifungice, antibiotice şi/sau antitermice, tratamentul chirurgical al abceselor localizate, administrare de imunoglobuline specifice, anticorpi monoclonali specifici – omalizumab. Acesta este un agent imunoterapeutic folosit ca anti-IgE, dar cu eficacitate terapeutică mai mult în astmul sever, la pacienţii sub 12 ani, cu boală alergică diagnosticată şi controlată insuficient cu corticosteroizi inhalatori, decât în sindromul hiper-IgE.
Prognosticul este grevat de apariţia infecţiilor pulmonare recurente şi cutanate cu potenţial de sepsis şi de complicaţiile pneumatocelelor.
Complicaţiile ce pot apărea sunt infecţii severe stafilococice cutanate şi pulmonare, infecţii în sfera stomatologică, infecţii pentru care în mod obişnuit se face imunoterapie prin vaccin cu organisme vii-atenuate (BCG, rubeolă, rujeolă, febră galbenă, rotavirus, gripă, poliomielită). Alte probleme ce pot apărea din cauza statusului neurologic pot fi scolioze cu evoluţie rapidă şi apariţia de malignităţi, preponderent limfoame.
Particularitatea cazului
Cazul prezentat evocă un aspect tipic de sindrom hiper-IgE, caracterizat prin prezenţa chistului aerian pulmonar, a dentiţiei duble şi a abceselor cutanate. Ca particularitate, HIES este asociat sau complicat cu o meningită soldată cu sechele neurologice grave. Meningita de la vârsta de trei luni a fost o complicaţie a deficitului imun din cadrul acestei entităţi sau o simplă faţetă a bolii.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
- Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. 1966; May 7.1(7445):1013-5. (Medline)
- Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972; Jan.49(1):59-70. (Medline)
- Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005; Feb.203:244-50. (Medline)
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007; Aug 30.448(7157):1058-62. (Medline)
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007; Oct 18.357(16):1608-19. (Medline)
- Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci. 2012; Feb.1250:25-32. (Medline)
- Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore). 2012; Jul.91(4):e1-19. (Medline)
- Minegishi Y, Saito M. Molecular mechanisms of the immunological abnormalities in hyper-IgE syndrome. Ann N Y Acad Sci. 2011; Dec.1246:34-40. (Medline)
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008; Apr 10.452(7188):773-6. (Medline) (Full Text)
- Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V. Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol. 2007; May 119(5):1234-40. (Medline)
- Leonard GD, Posadas E, Herrmann PC, Anderson VL, Jaffe ES, Holland SM. Non-Hodgkin’s lymphoma in Job’s syndrome: a case report and literature review. Leuk Lymphoma. 2004; Dec.45(12):2521-5. (Medline)
- O’Connell AC, Puck JM, Grimbacher B, Facchetti F, Majorana A, Gallin JI, et al. Delayed eruption of permanent teeth in hyperimmunoglobulinemia E recurrent infection syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000; Feb.89(2):177-85. (Medline)
- Ling JC, Freeman AF, Gharib AM, Arai AE, Lederman RJ, Rosing DR. Coronary artery aneurysms in patients with hyper IgE recurrent infection syndrome. Clin Immunol. 2007; Mar.122(3):255-8. (Medline)
- Freeman AF, Collura-Burke CJ, Patronas NJ, et al. Brain abnormalities in patients with hyperimmunoglobulin E syndrome. Pediatrics. 2007; May.119(5):e1121-5. (Medline)
- Ozcan E, Notarangelo LD, Geha RS. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol. 2008; Dec.122(6):1054-62;quiz 1063-4. (Medline)
- Jonczyk-Potoczna K, Szczawinska-Poplonyk A, Warzywoda M, Breborowicz A, Pawlak B. Hyper Ig E syndrome (Job syndrome, HIES) - radiological images of pulmonary complications on the basis of three cases. Pol J Radiol. 2012; Apr.77(2):69-72. (Medline)

Tuberculoza pulmonară − o provocare la vârsta pediatrică
Roxana-Iuliana Chihaia, Evelina Moraru, Mihai-Ioan Colţa, Bogdan A. Stana
Tuberculoza pulmonară încă reprezintă o provocare pentru medicul practician, intrând în diagnosticul diferenţial al oricărei opacităţi pulmonare. Deşi vaccinarea este realizată corect în România, totuşi există un număr r...
Consangvinitatea − la limita dintre etic şi patologic
Irina-Mihaela Ciomagă, Emma Oana Horhotă, Mădălina Mardare, Elena Ţarcă, Aniela Rugină, Doina Mihăilă, Petru Plămădeală, Nicolai Nistor
Căsătoriile consangvine sunt definite ca fiind acele căsătorii în care partenerii au un ascendent comun. Consangvinitatea pune probleme atât de natură etică, deoarece este considerată imorală,...
Evaluarea cazurilor de intoxicaţii cu insecticide inhibitorii de colinesterază la copil: studiu retrospectiv pe 7 ani
Nicolai Nistor, Cristina Jităreanu, Tamara Solange Roşu, Ştefana Dupa, Dana Elena Mîndru, Irina-Mihaela Ciomagă
În acest studiu retrospectiv au fost evaluaţi 87 de copii internaţi în Centrul regional de toxicologie al Spitalului Clinic de U...
Evaluarea cazurilor de intoxicaţii cu insecticide inhibitorii de colinesterază la copil: studiu retrospectiv pe 7 ani
Nicolai Nistor, Cristina Jităreanu, Tamara Solange Roşu, Ştefana Dupa, Dana Elena Mîndru, Irina-Mihaela Ciomagă
În acest studiu retrospectiv au fost evaluaţi 87 de copii internaţi în Centrul regional de toxicologie al Spitalului Clinic de U...
Implicaţii imediate şi la distanţă într-un caz de atrezie esofagiană operată
G.I. Criscov, O.I. Bărbuţă, Elena Ţarcă, Bogdan A. Stana
Atrezia esofagiană reprezintă discontinuitatea congenitală a esofagului, cu consecinţe digestive, respiratorii, nutriţionale şi infecţioase. Recunoaşterea precoce a acestei patologii şi corecţia chirurgicală pot îmbunătă...