Neuromuscular diseases are disorders of the motor unit (the basic functional and anatomical organization of neurons and muscle fibers). Neuromuscular diseases involve pathologic processes that primarily are directed against, and are limited to, one or more subdivisions of the motor unit.
Each motor unit is composed of a single motoneuron, and a variable number of skeletal myofibers innervated by a single motoneuron. The essential components of each motor unit include:
1. A motoneuron consisting of its cell body (located within the CNS, either in the cranial nerve nuclei of the brainstem, or in the ventral horns of grey matter in the spinal cord) and its peripheral axon, supported by Schwann cells;
2. Neuromuscular junctions;
3. Myofibers innervated by the motoneuron.
Classification of neuromuscular disorders
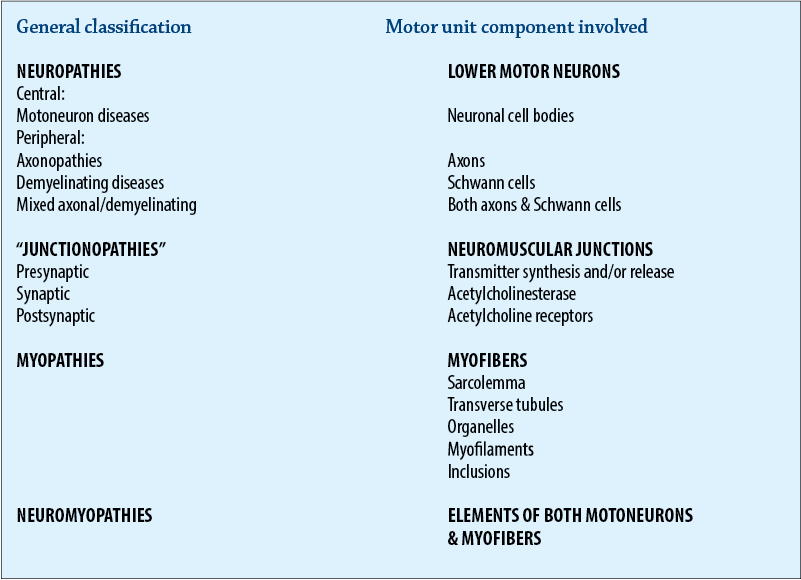
A useful classification scheme of neuromuscular diseases is based on the anatomic motor unit components that are primarily involved in the pathogenesis of the muscle weakness. Using this classification, neuromuscular diseases are broadly subdivided into:
1. Neuropathies - disorders of the neuron, its cell body, axon, and/or Schwann cells (myelin)
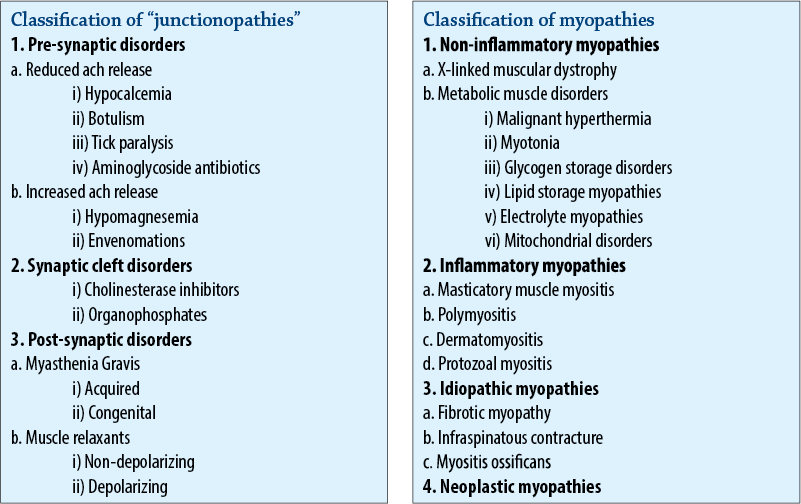
2. “Junctionopathies” - disorders of neuromuscular junctions
3. Myopathies - disorders of muscle fibers
4. Neuromyopathies - disorders of both the neurons and muscle fibers.
Clinical signs of neuromuscular disorders
1. Generalized or localized muscular weakness
2. Functional manifestations:
Paresis/paralysis
Gait abnormalities
Exercise related weakness
Dysphagia
Regurgitation
Dyspnea
Dysphonia.
3. Physical manifestations:
Muscle atrophy/hypotrophy
Muscle hypertrophy
Skeletal deformities.
Cervical ventroflexion is a dramatic sign of generalized neuromuscular weakness in cats. The chin usually rests near the thoracic inlet, with the eyes positioned dorsally to maintain a straight-ahead gaze. Other common physical examination findings are a slight protrusion of the dorsal aspects of the scapulae when weight is placed on thoracic limbs, and a stiff thoracic limb gait. A crouched, wide-based stance is often seen in pelvic limbs. Possible causes to consider for this posture are: subacute or chronic organophosphate toxicity, potassium-depletion myopathy, thiamine-responsive neuromuscular weakness, hyperthyroidism, immune-mediated (idiopathic) polymyositis, myasthenia gravis, polyneuropathy, hypernatremic polymyopathy, ammonium chloride toxicity, hereditary myopathies (Burmese, Devon Rex), hypocalcemia, and portosystemic encephalopathy.
Diagnosis of neuromuscular disorders
Establishing a diagnosis requires an informed and coordinated approach to defining a problem list through associations and direct observations (i.e., a diagnostic plan).
1. Signalment, history, physical and neurological examinations
Signalment: species, breed, age, sex, use.
History: congenital/acquired, course of complaint, response to treatment, exposure to toxins, etc.
Findings: presence and distribution of abnormal findings on physical and neurological examinations.
2. Minimum data base
Minimum data base: CBC, serum biochemistry panel, urinalysis, thoracic radiographs, and abdominal ultrasound.
Measurement of muscle specific serum enzymes such as creatine kinase (CK), as well as aspartate aminotransferase (AST), and lactic dehydrogenase (LDH), is very helpful in identifying neuromuscular disorders in which myonecrosis is a principal pathologic feature. Elevated serum enzyme activities help to differentiate myopathies from other neuromuscular disorders. Also immunologic procedures for the detection of myoglobin are becoming available, and should be a sensitive means of detecting myolysis in the future.
3. Specific diagnostic tests - electrodiagnostic testing
4. Specific diagnostic tests - nerve and muscle biopsy examination
Histopathological features of neuropathies
General features
Angular Atrophy. The hallmark of denervation consists of characteristic patterns of myofiber atrophy. Myonecrosis is an uncommon reaction to denervation. With minimal denervation, in which only a few myofibers are denervated, the denervated myofibers undergo atrophy and these atrophied myofibers tend to be angular in appearance. These angular atrophied type 1 and type 2 myofibers (“angular atrophy points to denervation”) tend to be scattered throughout the section and they are present in many fasciculi. It appears that this sign of denervation takes several weeks to develop after the denervating event.
Specific neuropathies
1. Motoneuron disorders
Hereditary and acquired motoneuron disorders have been described in dogs (spinal muscular atrophy of Brittany spaniels), horses (equine motoneuron disease), and cattle.
2. Peripheral neuropathies
a. Mononeuropathies
b. Multiple mononeuropathies
c. Polyneuropathies.
Polyneuropathies represent the single largest group of neuromuscular disorders. Many are idiopathic (i.e., our diagnostic skills are insufficient to establish the etiology). Disorders include: acute and recurrent polyradiculoneuritis, metabolic (diabetes, hypothyroidism, hypoadreno-corticism, hyperinsulinism), toxic, lysosomal storage disorders (e.g., Krabbe’s globoid cell leukodystrophy or galacto-cerebrosidase deficiency, and Niemann-Pick disease or sphingomyelinase deficiency).
3. Undetermined.
Histopathological features of “junctionopathies”
General features
Histopathological changes usually are absent or non-specific in disorders of the neuromuscular junction. Diagnoses usually are based on biochemical, immunological, toxicological, electrodiagnostic, and/or immunological testing.
Histopathological features of myopathies
General features
In small animals, myopathies are relatively uncommon and encountered less frequently than neuropathies and junctionopathies, while the converse generally is true in large animals.
Myopathies may be subdivided into non-inflammatory and inflammatory myopathies.
Non-inflammatory myopathies
The histopathological changes in non-inflammatory myopathies usually involve the spectrum of myonecrosis, phagocytosis, and regeneration, in which the degree of cellular infiltration is proportional to the extent of myonecrosis present, and its distribution is largely limited to necrotic fibers.
Macrophages constitute the principal cell type. Central nuclei are common.
In chronic myopathies there may be a mixture of atrophied and hypertrophied fibers, with their morphology being more “anguloid” than angular, and increased endomysial connective tissue.
Occasionally necrotic fibers may be calcified.
All these changes are relatively non-specific and secondary to agents that result in myonecrosis. In metabolic myopathies the fibers frequently contain storage products that appear as vacuoles.
Inflammatory myopathies
The inflammatory myopathies possess many of the changes described for non-inflammatory myopathies. However, in addition, they are characterized by a disproportionate number of infiltrating cells that may include lymphocytes/plasma cells, polymorphonuclear leukocytes and/or eosinophils in addition to macrophages. In these disorders, the cellular infiltrates comprise an integral part of the disorder’s pathogenesis and not merely a secondary response to cell death.
The infiltrating cells often have a perivascular distribution.
Identification and characterization of the infiltrating cell type assists in the definition and recognition of these disorders.
The use of the term “myositis” is reserved for inflammatory myopathies. Inflammatory myopathies may be caused by infectious or immune-mediated disorders.

Recomandări terapeutice pentru traumatisme cerebrale
Richard A. LeCouteur
Primary or biomechanical injury is the injury to the brain tissue from direct trauma and the forces applied to the brain at impact. ...
Afecțiuni transmise prin sânge la feline
Michael R. Lappin
A number of feline blood borne agents have been grown, amplified or have induced serum antibodies in the serum of cats with clinical signs like fever. The purpose of this comprehensive review is to provide an update on the diagnosis and management of fever associated with Anaplasma phagocytophilum, Bartonella...
Afecțiuni ale tractului respirator inferior la feline
Michael R. Lappin
Bronchial obstruction can develop due to inflammatory infiltrates (eosinophils, neutrophils, or macrophages) or hypertrophy of bronchial tissues. The result is obstructive airway disease. Obstructive airway disease is characterized by increased airway resistance with resultant expiratory dyspnea. ...