Asthma: from complex pathophysiology to histological changes
Astmul bronşic: de la patogenie complexă la modificări histologice
Abstract
Asthma is the most common chronic lung disorder and affects approximately 15-20% of the population in developed countries, whereas the rate is around 2-4% in less developed countries. The incidence rates are much higher in children, with roughly 40% of children reporting having a wheezing episode which can turn at some point into asthma. Understanding pathogenesis can help to better treat those patients. The release of these inflammatory mediators is the primary cause of the key feature of asthma – chronic airway inflammation. This in turn leads to the airway remodeling phenomenon, which is seen by the thickened airway walls and hence the narrowing of the air passages, ultimately having a profound impact on the course and chronicity of the condition.Keywords
asthmachildreninflammationRezumat
Astmul este cea mai frecventă afecţiune pulmonară cronică şi afectează aproximativ 15-20% din populaţie în ţările dezvoltate, în timp ce rata este de aproximativ 2-4% în ţările mai puţin dezvoltate. Ratele de incidenţă sunt mult mai mari la copii, aproximativ 40% dintre aceştia declarând că au avut un episod de respiraţie şuierătoare care se poate transforma la un moment dat în astm. Înţelegerea patogenezei poate ajuta la o mai bună tratare a acestor pacienţi. Eliberarea mediatorilor inflamatori este cauza principală a inflamaţiei cronice a căilor respiratorii. Aceasta, la rândul său, conduce la fenomenul de remodelare a căilor respiratorii, care se manifestă prin îngroşarea pereţilor căilor respiratorii şi, prin urmare, prin îngustarea acestora, având în cele din urmă un impact profund asupra evoluţiei şi cronicizării afecţiunii.Cuvinte Cheie
astmcopiiinflamaţieThe major factors contributing to asthma are atopy (genetic predisposition to type I hypersensitivity), acute or chronic inflammation of the respiratory tract and bronchial hyperresponsiveness to various stimuli. Thus, we can subclassify asthma as atopic or nonatopic. In both types, episodes of bronchospasm can be triggered by different exposures, such as respiratory infections (mainly of viral etiology), certain respiratory irritants, cold air, stress or physical exertion. There are also different patterns of inflammation: eosinophilic (most common), neutrophilic, mixed and pauci-granulocytic, which are associated with different etiologies, with immune pathologies or occur in response to some treatments(1).
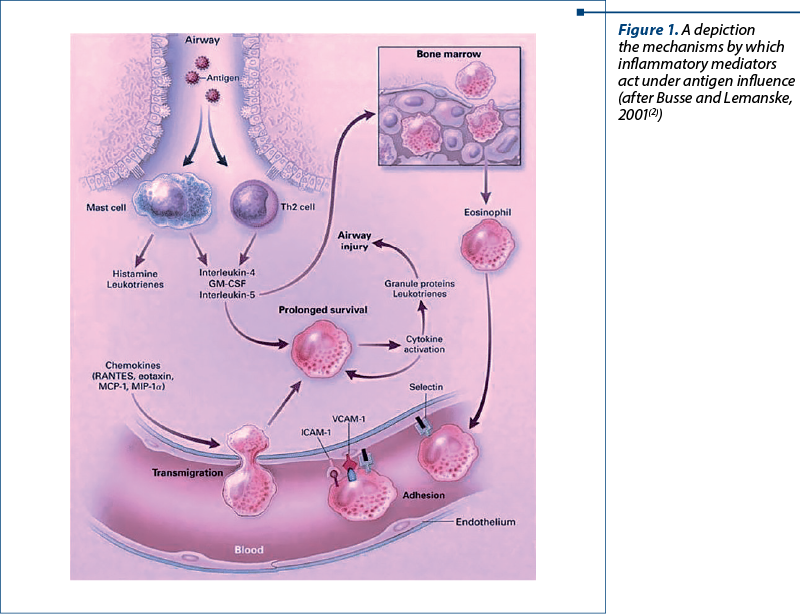
The classical atopic form is associated with exaggerated activation of Th2 cells. Cytokines produced by Th2 are responsible for most features of atopic asthma: IL-4 and IL-13 stimulate IgE production, IL-5 activates eosinophils, and IL-13 stimulates excessive mucus production. IgE engulfs mast cells in the submucosa, which when exposed to an allergen will release their granulocyte contents and secrete cytokines as well as other mediators (Figure 1). These mast cell-derived mediators produce two types of reactions: an early (immediate) reaction and a late reaction (Figure 2). The early reaction is dominated by bronchoconstriction, increased mucus production and vasodilation. Bronchoconstriction is triggered by mediators released by mast cells (histamine, prostaglandin D2, leukotrienes LTC4, D4, E4) as well as by a series of neural reflex pathways. The late reaction is inflammatory in nature. Inflammatory mediators stimulate epithelial cells to produce chemokines (eotaxin – a chemotactic agent and eosinophil activator) which cause the recruitment of Th2, eosinophils and other leukocytes, which will amplify the inflammatory reaction. Repeated episodes of inflammation will lead to a series of changes in the bronchiolar wall; thus, a “remodeling” of the airways occurs. These changes include hypertrophy of bronchiolar smooth muscle and mucus-secreting glands, increased vascularization and the appearance of collagen deposits at the subepithelial level(1,3).
Bronchial asthma tends to occur as a hereditary condition, but the role of genetics in asthmatic pathology is very complex and still not fully understood. There are studies that have identified a number of genes that are associated with an increased risk of developing asthma (genes that convert the IL-4 receptor – which are clearly involved in the pathogenesis of the disease)(4).
Throughout the evolutionary increasing knowledge of asthma, a paradigm shift has been seen in certain scientific fields, which have provoked a deeper evaluation of the key role inflammatory mediators play in asthma pathophysiology. To compound this, the airway remodeling processes, which act to thicken the walls of the airways, further narrow the airway and add to the chronicity of the disease.
Broadly speaking, the vast majority of asthma cases begin in the juvenile childhood ages, as a result of inhaling allergens commonly found in a home or in an environmental setting. Allergens such as dust mites, pollen and animal dander provoke the proliferative phases of T-helper 2 cells, as mentioned earlier, which in turn produce and secrete the inflammatory mediators which infiltrate, namely the interleukins. The integral mechanism to the pathophysiological process of asthma is generally agreed to be airway inflammation, and the releasing of the mediators of the process is the fundamental start point.
Often, the first step is the uptake of the allergens by dendritic cells, causing allergic sensitization to occur. The dendritic cells then function to present the allergens to naïve T helper cells, which activate these T cells. Recent studies and advancements have, on a sidenote, implicated the roles Th17 and Th9 cells also play in the disease process, by also stimulating certain interleukins (17 and 22), which also enhance the inflammatory process, as well as promoting a contractile process from the smooth muscle.
Several functional modifications occur during the asthma development, all contributing to the overall airflow limitation causing an obstructive disease process. The changes are as follows:
-
Bronchoconstriction – this process compounds the narrowing airway, further reducing the potential for external outflow. Often present during exacerbations, the contraction of bronchial smooth muscle is a rapid response in asthmatics when exposed to irritant allergens. Once inhaled, the allergens act by causing inflammatory mediator release from allergen-related immunoglobulin, IgE. The major bulk of the mediators are released by mast cells, and include histamine, leukotrienes, prostaglandins and tryptase, all of which are directly involved in smooth muscle contraction. The bronchoconstriction process is not limited to just allergen inhalation, as certain pharmacological compounds, especially NSAIDs and aspirin, also lead to some degree of airflow obstruction, by activating a different, non-IgE response(5). The limitation to airflow has also been seen when exposure to other factors, such as exercise or cold air, occur, perpetuating another route the disease can be caused by.
-
Airway oedema – as disease progression occurs towards more advanced stages, the already present bronchoconstriction is accompanied by other factors which further contribute to the limited airflow. Oedema development, along with excessive mucosal secretion which lead to mucous plug formations, and smooth muscle manifestations add more dimensions to the complexity of the disease.
-
Airway hyperresponsiveness – the degree to which the exaggeration of a response from the bronchi and their constriction occurs is a major factor, as there is correlation between this and the severity to which the disease is manifested in an individual.
-
Airway remodeling – as it was mentioned earlier, the structural modifications of the airway wall are often only partially reversible, and are frequently associated with an ongoing diminishing lung function. The remodeling effect produces alterations which enhance the degree to which airflow obstruction occurs, and subsequently leave the patient with a depreciated therapeutic response(6).
What is apparent throughout all this discussion is the centrality of inflammation to asthma pathogenesis, pathophysiology and symptomatology. By its definition, the inflammation involves several different types of cell and inflammatory mediators to produce the clinical effect of bronchial inflammation, airway obstruction and episodic coughs, associated with wheezing and dyspnea. A breakdown of the main cells involved in inducing and evoking an inflammatory response follows.
-
Lymphocytes – the advancements in understanding the inflammatory process in asthma was in part aided by increased knowledge pertaining to the descriptions and types of lymphocytes. The two main subtypes of this category of mediator are the T helper 1 and T helper 2 cells, each having unique effects on the airway as a whole. With animal studies performed, a shift was seen in the human population, with more weight placed on T helper 2 cytokines producing the heavily eosinophilic response characteristic of asthma(7). This excess eosinophil infiltration may be due to the cytokines produced by stimulated T helper 2 cells, namely the interleukins 4, 5 and 13 manufacturing excess IgE. Along with these phenomena, a decrease of one lymphocyte subset, the regulatory T cells, occurs. The function of this group is to suppress and inhibit the T helper 2 cells, so it follows that their reduction further exacerbates the inflammatory mechanism. A simultaneous increase in natural killer (NK) cells is also seen, and these cells release even more both T helper 1 and 2 cells. The influence of lymphocytes is clearly huge on the overall extent to which asthma can manifest itself and remodeling can occur.
-
Mast cells – primarily located in mucosal areas, these cells release substances which collectively come under the category of “bronchoconstrictor mediators”, and include leukotrienes, histamine and prostaglandins, with “prostaglandin D” being the main one(8). Although the main allergen activation occurs via IgE receptors, as mentioned previously, mast cells also play an important role as certain osmotic stimuli tend to cause their activation, which is often visible in conditions of “exercise-induced bronchospasm”. The smooth muscle response is also triggered by mast cell presence in the muscle itself.
-
Eosinophils – most, if not all people afflicted with asthma have enhanced eosinophil content in their airways. Their nature being that of enzyme containment which trigger inflammatory responses, and the fact they both present many cytokines and produce leukotrienes means that the level of their excess correlates to the severity degree of the disease itself. Their influence is also clear when seeing reductions in their amount, along with improved clinical condition when corticosteroid therapy is administered.
-
Neutrophils – the role of neutrophils is still not fully understood; however, their presence in exaggerated quantities is seen in the airways and sputum of people confirmed to have asthma.
-
Dendritic cells – by functioning as antigen presenting cells, dendritic cells communicate with the allergens at the level of the airway, before migrating lymphatically to reach the regulatory cells, with the end result being the stimulation of naïve T cells to proliferate and produce T helper 2 cells(9).
-
Macrophages – allergens acting via IgE receptors activate macrophages, which act by also increasing the production and release of cytokines and other such inflammatory mediators, exacerbating the airways immune response.
The smooth muscle content of the airway acts in two ways: as a target of asthma response by increasing the contraction to degree, ergo limiting outflow and, secondly, directly contributing to asthma response through the release of its own inflammatory molecules and also through the hypertrophic and hyperplastic mechanisms mentioned before. The key compounds involved in airway smooth muscle are acetylcholine and methacholine, which activate the compound phospholipase C. This in turn acts by forming the inositol triphosphate substance, which is crucial as this is the substance that allows the release of calcium ions from the muscles’ sarcoplasmic reticulum calcium ion storage. The released calcium is then able to bind to calmodulin, causing the “calcium-calmodulin complex” to be formed. The complex then acts by activating the enzyme MLC kinase to be able to phosphorylate regulatory MLCs, therefore forming phosphorylated-MLC. The final step is the activation of actin and myosin cross-bridges, which are then able to shorten and cause smooth muscle contraction(2).
As Figure 1 depicts, the antigens when inhaled activate the mast and T helper 2 cells at the airway level. Consequently, the production of inflammatory mediators is done, with chemicals such as histamine and leukotrienes undergoing mass production, as well as cytokines, including the interleukins 4 and 5. IL-5 redirects to bone marrow, differentiating the eosinophils. These mature forms of eosinophils then are able to interact with selectins, and travel through the lung space and attach to the endothelium, via integrin binding to adhesion proteins. The main adhesion proteins involved in this process are vascular-cell adhesion molecule 1 and intercellular adhesion molecule 1. Under the direction of the chemo and cytokine mediators, their overall chances of thriving are enhanced by the actions of IL-4 and granulocyte-macrophage colony-stimulating factor. Once activated, the eosinophils are in turn able to secrete inflammatory mediators of their own to attack and damage airway tissue, as well as producing granulocyte-macrophage colony-stimulating factor of its own to further aid its survival and potentiate the inflammatory process. Other contributing compounds in this process includes monocyte chemotactic protein and macrophage inflammatory protein(2).
As mentioned throughout, the role of inflammatory mediators is widespread, with some examples following.
-
Chemokines – these mediators are vital in inducing inflammatory cells into airways and are present abundantly in epithelial cells.
-
Cytokines – these mediators are involved in the response to asthma and the amount of these cytokines determines how severe the response is to this reaction. IL-5 is necessary for the differentiation of eosinophil and is derived from Th-2. IL-4 is also derived from Th-2 and is involved in the differentiation of Th-2 cells. IL-13 is needed to produce IgE. Tumor necrosis factor and interleukin-1b further add to the magnitude of the inflammatory response, along with the ever-present granulocyte-macrophage colony-stimulating factor aiding its lifespan in the airway
-
Leukotrienes – in their role as bronchoconstrictors, they are unique in that their suppression is directly related to an enhanced lung function and overall clinical presentation.
-
Immunoglobulin E – as the immunological compound overseeing the activation of hypersensitivity reactions, it is central to the pathogenic process as well as the inflammatory one. Its functionality is permitted via its attachment to its respective receptors. Mast cells represent a large store of receptors, and when stimulated by the presence of antigens, the secretion of a plethora of mediators act to produce episodic bronchospasms, along with cytokine release to further propel the existing inflammation. Dendritic cells, lymphocytes and basophils also contain IgE receptors, so the responsibility does not fall solely on mast cells. Studies evaluating suppression of IgE by utilizing monoclonal antibody techniques have consistently demonstrated the enhanced efficacy and effectiveness of asthma therapy regimens when the circulating IgE levels are diminished(6).
Histopathology of asthma
When discussing the histopathological aspects and manifestations of asthma on a microscopic level, it is important to reiterate the “hallmarks” of the disease as a whole, which include the partial reversibility of an obstructed airflow, the hyperreactive bronchus, and the chronic inflammation of the airway as a whole. Classic medical literature has described for over a century the idea of an inflammatory process, also outlining a relative lack of effect on the parenchyma. Further evidence pertaining to the histological specifics was shown in the early part of the 20th century, claiming that lungs undergo a hyperinflation as a result of higher levels of bronchial and peripheral mucosal plugging, while sparing the parenchyma, as said before. The mucosal plugging phenomenon involves an accumulation of debris from necrotic cells, along with general inflammatory cells such as lymphocytes and eosinophils. Exudations from plasma proteins and mucin secreted by goblet cells also perpetuate the plugging process. Furthermore, a metaplastic process is seen involving squamous cells and goblet cells, indicating a degree of epithelial repair occurring. The basement membrane, as spoken about extensively earlier, thickens in correlation to an accumulated matter of extracellular matrix components. The airway thickening is due to inflammatory infiltrate by the respective cell types mentioned, but a discovery promoting more inquisition is the predominance of neutrophils being a key aspect of the clinical morphology of a sudden-onset fatal type of asthma disease(10).
Modern advancements have given a pathway to a better assessment of the microscopic modifications that occur. Bronchoalveolar lavage techniques have made it possible to be able to sample lung tissue from asthmatic patients, and studies done via lavage techniques have demonstrated the role of the cytokines produced by T-helper 2 cells in asthma pathogenesis, specifically that of inflammatory regulation by way of IL-13 in asthma(11).
Extracellular depositions have added a lot of impetus to the airway remodeling idea, cementing the belief that inflammation is not the sole contributor to the progression of asthma. While injured, the damaged epithelium is a perpetual stimulus for remodeling to occur, and studies of cell cultures looking into how the epithelium of the bronchi behave with myofibroblasts have backed these claims(6).
The thickening of basement membranes, and therefore the airway as a whole, is certainly larger in asthmatic patients, at least when comparisons are done to normal healthy people, but the fact that the thickness is greater as severity of disease increases is an important distinction to make. Increased mass, mainly of smooth muscle, but also of mucous glands is the primary driving force behind this increased thickness, and thus airflow limitation occurs, further worsened by increased secretion from the hypertrophied mucous glands, along with inflammatory exudate secreted. A somewhat surprising finding from several studies, however, is the induction of remodeling purely by the fore generated by bronchial constriction(12).
Medical research centers based in both the University of Pittsburgh’s asthma institute and the same institutions medical center undertook a fascinating study over an 8-year period, from 2007 to 2015, involving taking surgical lung biopsies from twenty-nine patients diagnosed with “severe/therapy-resistant asthma”, with these meeting the definitions determined by both the ERS (European Respiratory Society) and ATS (American Thoracic Society), respectively(13). The need for biopsies was indicated due to the significant clinical severity of their conditions, and the fact they were refractive and not responsive to conventional therapeutic regimens, in order to potentially exclude “pseudo-asthma” conditions or other comorbidities.
The study concluded with clinical, radiologic and histologic parameters of the patients with confirmed severe asthma conditions, by way of video-assisted thoracoscopic surgery lung biopsy. Corroborations were made between clinical aspects and pathological findings, particularly the presence of autoimmune disease, which appeared in just over 50% of cases. Ten of the biopsies portrayed minor airway modifications, with nineteen demonstrating asthmatic granulomatosis, which could be confirmed by seeing an asthmatic bronchiolitis exacerbated by alveolar septal mononuclear infiltrates. In patients showing no granulomatosis, the intensity of small-grade airway injury was higher, along with BM thickening and infiltrating neutrophils. The autoimmune disease patients conversely had an eosinophilic infiltration predominance into the parenchyma, along with being more responsive to non-steroidal immunosuppressive treatment. The histology of the studied patients showed a broad spectrum of pathologies.
The histology of the subjects was based from wedge biopsy samples from the lung parenchyma of a minimum of two lung lobes. The biopsy samples were fixed using formalin and paraffin wax, before being stained using hematoxylin-eosin and Ziehl-Neelsen staining methods. The microscopic examination showed several key changes, such as mucous plugs, basement membrane thickening, bronchiolectasis, goblet cell metaplasia, hypertrophied smooth muscle, aggregation of lymphoid derivatives and in some cases granuloma presence was seen, as well as morphological descriptions and localities noted.
Further examination of the histologic changes indicated that hypertrophy of smooth muscle was present in all biopsied specimens. The changes were not limited to the larger bronchi, with the smaller, distal airways also manifesting these changes. All specimens also underwent a basement membrane thickening process, along with both mucous plugging and goblet cell metaplasia; however, fewer cases or a lesser degree of cases were present in autoimmune disease patients compared to granulomatous patients(13).
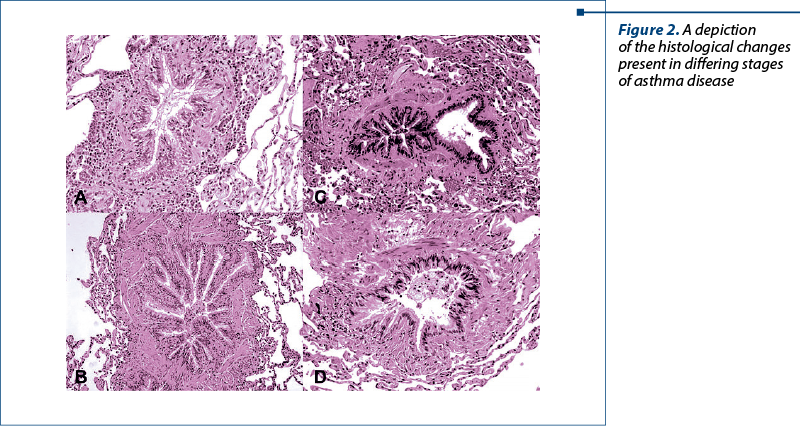
Figure 2 illustrates the morphological changes found in the smaller airways of these patients with severe or therapy-resistant forms of disease. Image A highlights the mucous plug formation, along with evidence of a metaplastic process of goblet cells, as well as inflammatory infiltrate, primarily being eosinophilic. An autoimmune concurrence is seen in image B. On the other hand, image C portrays the aspect of a severe asthma case with granulomatosis presence. Image D is also a granulomatous case, however differs due to having a concurrent autoimmune disease also. These visual representations also go some way to aiding visualization of mucous plugging as well as the ever-present smooth muscle hypertrophy(13).
The presence of inflammatory infiltrate was not limited to just the bronchi and smaller airways, as the parenchyma also showed evidence of being affected. Eosinophils were only found in granulomatous and autoimmune disease cases, but to a limited extent. On the contrary, neutrophils were seen in parenchyma in small numbers in only granulomatous cases, with the parenchyma being spared from neutrophilic infiltrate in regular severe cases and autoimmune cases. Lymphocytes, however, were ever-present in all types of studied disease, but with greater frequency in granulomatous and autoimmune subjects, seen by aggregations of lymphoid tissue throughout the parenchyma. The final morphologic change seen was all groups highlighting airspace organization to some extent.
Ziehl-Neelsen staining and Gomori methenamine staining techniques were performed to attempt to see any microbe invasion, however no bacterial or fungal species were discovered, yielding a negative result to the test. Other comorbid findings searched for included vasculitis, necrotic process, aspiration of foreign material, honeycombing, fibroblasts, autoimmune diseases (rheumatoid arthritis, SLE) and bronchitis/bronchiolitis. All of these additional findings reported negative results and were not found in any of the sampled specimens(13).
Conclusions
Physiopathogenic events in asthma are complex, leading to histological changes which correlate with disease severity and progression. Understanding those processes may help in better managing the disease.
Conflict of interests: The authors declare no conflict of interests.
Bibliografie
-
Paramothayan, S. Essential Respiratory Medicine (Essentials), 1st Edition, Wiley-Blackwell, 2019.
-
Busse WW, Lemanske RF. Advances in Immunology. N Engl J Med. 2001;344:350-62.
-
Berankova K, Uhlik J, Honkova L, Pohunek P. Structural changes in the bronchial mucosa of young children at risk of developing asthma. Pediatr Allergy Immunol. 2014;25:136–142. doi: 10.1111/pai.12119
-
Bao K, Reinhardt RL. The differential expression of IL-4 and IL-13 and its impact on type-2 immunity. Cytokine. 2015;75(1):25-37.
-
Stevenson DD, Szczeklik A. Clinical and pathologic perspectives on aspirin sensitivity and asthma. J Allergy Clin Immunol. 2006;118(4):773–86. quiz 787–8.
-
Holgate ST, Polosa R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet. 2006;368(9537):780–93.
-
Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815.
-
Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–86.
-
Kuipers H, Lambrecht BN. The interplay of dendritic cells, Th2 cells and regulatory T cells in asthma. Curr Opin Immunol. 2004;16(6):702–8
-
Hamid Q, Song Y, Kotsimbos TC, Minshall E, Bai TR, Hegele RG, et al. Inflammation of small airways in asthma. J Allergy Clin Immunol. 1997;100:44–51. 10.1016/S0091-6749(97)70193-3
-
Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalties, and eotaxin production. J Clin Invest. 1999;103:779–788. 10.1172/JCI5909
-
Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364 2006–2015. 10.1056/NEJMoa1014350
-
Trejo Bittar HE, Doberer D, Mehrad M, et al. Histologic Findings of Severe/Therapy-Resistant Asthma From Video-assisted Thoracoscopic Surgery Biopsies. Am J Surg Pathol. 2017;41(2):182-188. doi: 10.1097/PAS.0000000000000777.

Hepatitis B virus – a silent pathogen with many faces of chronic infection. Case report
Mara Midena Puiu, Alina Grama, Gabriel Benţa, Patricia Lorinţiu, Florina Larionesi, Claudia Simu, Tudor Lucian Pop
Hepatita cronică cu virus B este o cauză majoră de mortalitate şi morbiditate la nivel mondial, mai ales atunci când modalitatea de transmitere este verticală (de la mamă la făt) sau orizontală, în sp...
Diagnostic challenges in neonatal gynecomastia – case report
Alexandra Mirică, Laura Olăroiu, Alexandra Buică, Gabriel Coman, Diana-Loreta Păun, Ioana Anca Bădărău, Radu Mirică, Diana Monica Preda
Ginecomastia reprezintă dezvoltarea benignă a ţesutului mamar glandular la pacienţii de sex masculin. Mărirea în perioada neonatală a sânilor este o afecţiune frecventă datorată epuizării hormonilor...
Portal cavernoma in children – complications and evolution
Diana-Alexandra Borcău, Alina Grama, Simona Căinap, Claudia Simu, Patricia Lorinţiu, Gabriel Benţa, Bianca Raluca Mateescu, Mihaela Coşarcă, Tudor Lucian Pop
Tromboza de venă portă este o afecţiune rară, dar reprezintă o cauză importantă de hipertensiune portală în patologia pediatrică. ...
Portal cavernoma in children – complications and evolution
Diana-Alexandra Borcău, Alina Grama, Simona Căinap, Claudia Simu, Patricia Lorinţiu, Gabriel Benţa, Bianca Raluca Mateescu, Mihaela Coşarcă, Tudor Lucian Pop
Tromboza de venă portă este o afecţiune rară, dar reprezintă o cauză importantă de hipertensiune portală în patologia pediatrică. ...
Analiză clinică a malformațiilor arcului aortic: perspective din multiple studii de caz
Cosmin Diaconescu, Dana-Elena Mîndru, Bogdan A. Stana, Cristina Stoica, Alexandra Tincu, Tudor Tincu, Heidrun Adumitrăchioaiei, Alina-Costina Luca
Malformațiile arcului aortic sunt boli congenitale rare, care adesea se prezintă fără simptome, făcând dificilă documentarea lo...