Imunogenetica bolii Alzheimer: antigenul leucocitar uman
Immunogenetics of Alzheimer’s disease: the human leukocyte antigen
Abstract
Alzheimer’s disease (AD) is predicted to become the sixth leading cause of early death globally by 2040. Since the currently available treatment options are considered rather symptomatic, disease-modifying agents, mirroring a more in-depth understanding of the pathogenic mechanisms, are needed. There are some well-established pathogenic theories, but none of these are necessary, nor sufficient for pathogenesis. The inflammation pathogenic theory of the Alzheimer’s disease has been more and more studied for the last three decades, to try to fill the gap in the state of the art. One key player involved in the immune response of this chronic neuroinflammation is the human leukocyte antigen (HLA), the human equivalent of the major histocompatibility class. This paper reviews the current evidence for the involvement of the HLA system in the pathogenesis of Alzheimer’s disease.
Keywords
human leukocyte antigenHLAAlzheimer diseasedementiaimmunogeneticsinflammationRezumat
Se estimează că boala Alzheimer va ajunge a şasea cauză de deces prematur la nivel global până în anul 2040. Având în vedere că tratamentele actuale se consideră a fi doar simptomatice, sunt necesare terapii patogenetice care să reflecte o mai bună înţelegere a mecanismelor fiziopatologice ale bolii. Există câteva teorii fiziopatologice cunoscute, dar niciuna dintre acestea nu este nici necesară, nici suficientă pentru patogeneză. Teoria inflamaţiei este studiată în ultimele trei decade pentru a umple lacunele de cunoaştere privind patogeneza. În cadrul acestei neuroinflamaţii mediate de răspunsul imun, un rol major îl are antigenul leucocitar uman, echivalentul uman al complexului major de histocompatibilitate. Lucrarea de faţă analizează evidenţele privind implicarea antigenului leucocitar uman în patogeneza bolii Alzheimer.
Cuvinte Cheie
antigenul leucocitar umanHLAboala AlzheimerdemenţăimunogeneticăinflamaţieIntroduction
Alzheimer’s disease (AD), the most frequent major neurocognitive disorder, doubled in prevalence in the past 25 years, and it is predicted to become the sixth leading cause of early death globally by 2040(1). Being a major public health problem, it needs new predictive, preventive and curative measures. Since the currently available treatment options are considered rather symptomatic (i.e., cholinesterase inhibitors, NMDA receptor blockers)(2), disease-modifying agents, mirroring a more in-depth understanding of the pathogenic mechanisms, are needed(3,4).
Some well-established pathogenic theories are neuronal death and synaptic loss that result from the formation and accumulation of the extracellular amyloid plaques and the intraneuronal neurofibrillary tangles through the amyloid cascade and the hyperphosphorylation of tau-protein, starting from the amyloid precursor protein (APP). Mutations of the genes coding for the APP (chr 21), or for the proteases involved in the process (PRES1 – chr 14 and PRES2 – chr 1) proved to be disease-causing mutations(5,6), but are considered to be rather rare, accounting for the familial, autosomal dominant form of AD – i.e., the early-onset form, that represents less than 1% of all cases of AD(8).
APOE e4 allele (chr 19) was found to be the most important genetic risk factor for late-onset AD (LOAD), although the pathophysiological pathway remains largely unknown. APOE is a major apolipoprotein expressed in the brain, playing a role in mediating synaptogenesis, synaptic plasticity and neuroinflammation(7); however, mutations can be found only in 50% of patients(8).
We can therefore conclude that none of these already established theories are necessary, nor sufficient for AD pathogenesis(9,10).
The extracellular amyloid plaques and the intraneuronal neurofibrillary tangles deposits in the brain act as antigens. They activate microglia, the brain’s resident macrophage, and astrocytes, initiating the inflammation cascade(11). This results in the invasion of peripheral monocytes, neutrophils and CD8 T cells disrupting the brain blood barrier, a disruption that has been found to be a critical component of AD pathogenesis(12). The macrophage phagocytic dysfunction results in the apoptosis of the macrophage, further accumulating the amyloid deposits(13). The released proinflammatory cytokines (IL-18, IL-1b, TNF-a, ROS, NO etc.(15)) can increase the production of APP and amplify the amyloid cascade, thus entering a self-reinforcing vicious circle(14,16,17) of chronic inflammation, referred to as abnormal inflammatory status(13), that leads to synapse loss(18).
The inflammation pathogenic theory of the Alzheimer’s disease has been studied for the last three decades to try to fill the gap in the state of the art(19,20), some authors even stating that it begins decades before AD clinical onset(21).
It is supported by observational studies that found increased infiltrations of monocytes/macrophages, lymphocytes and T cells in the brain of AD animal models and humans(22,23). It is also supported by epidemiological studies showing a low incidence of AD in patients with long-term anti-inflammatory treatment(14,15,24), although it could not be proven in clinical trials(14). Furthermore, it is supported by shared genetic variants between some autoimmune disease and AD(25,26). Recent genome-wide association studies identified more than 30 genetic loci for AD, many related to immune response (e.g., CD33, INPP5D, CLU, TREM2, CR1, SPI1, ABCA7, EPHA1, MS4As, INPP5D, MEF2C, HLA-DRB5-DRB1)(27,28).
One key player involved in the immune response of this chronic neuroinflammation is the human leukocyte antigen (HLA), the human equivalent of the major histocompatibility class (MHC). The amyloid plaques that accumulate in the AD patients’ brain are internalized by the microglia and processed into pieces that, in combination with a specific HLA class I/II molecule, are presented to the T lymphocytes(29). The upregulation of HLA class II antigens is now accepted as a marker of the activated microglia(30). The resulted combination also stimulates B lymphocytes to secrete antibodies against amyloid peptides. Thus, MHC is a co-stimulatory molecule in the neuroinflammation cascade, one branch of the AD pathophysiology(29).
This paper reviews the current evidence for the involvement of the HLA system in the pathogenesis of Alzheimer’s disease. We searched the PubMed database using the MeSH terms “HLA” and “Alzheimer’s disease” for anytime published, full-text English written articles. Out of the 375 results given, we included the relevant once for the aim of this review.
The human leukocyte antigen system
The human leukocyte antigen (chr 6p21), first mapped in 1993(31) and 1999(32), is the densest region of the human genome, with more than 200 genes(33,34) and approximately 3.5 million base pairs(35). Its main function is the presentation of short peptides to specialized immune cells, playing a key role in innate and adaptative immunity.
It is divided into five subregions with different functions (Table 1)(26,33,35).
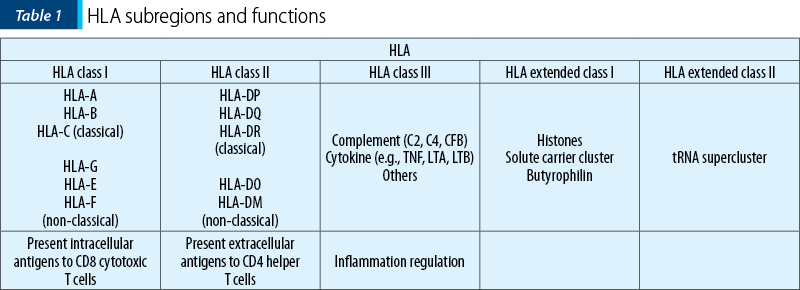
It is one of the most polymorphic regions in the human genome, up to date, 26,889 HLA alleles being named (19,578 class I and 7,302 class II)(36).
These code for a and b polypeptide chains that form an antigen binding groove: 2 a for HLA class I, a and b for HLA class II(34). Change in even one amino acid results in different antigen binding groove, thus a different HLA antigen that can bind a different pathogen(34). The match between the pathogen and the HLA groove is essential for the immune response(37,38). In the absence of a match, the pathogen can persist, causing inflammation, cell damage and autoimmunity(37). Therefore, it is likely that the polymorphism of this region represents an evolutionary adaption to the multitude of pathogens faced(34).
HLA class I and AD
In the central nervous system, HLA class I are highly expressed in the microglia and endothelial cells, lower levels being expressed in astrocytes, oligodendrocytes and neurons (where they play a role in synaptic plasticity)(39).
A great amount of research focused on HLA-A, especially on its most common allele HLA-A2, trying to associate it with AD pathogenesis or onset age. The results were highly inconsistent over more than two decades(40-50), probably due to small sample size, different HLA typing methods, participants’ ethnicity or study designs. More recent studies focusing on HLA-A SNPs tried to associate them with atrophy of cortical brain structures (rs9260168 and rs3823342 with left para-hippocampus, as seen on MRI), or subcortical structures (rs76475517 with amygdala). The study sample was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and included 812 participants (281 health controls, 483 participants in the mild cognitive impairment stage, and 48 AD participants), but the results were at a marginal significance (Pc=0.054)(51).
HLA-B and HLA-C are relatively understudied. HLA-B7 and HLA-Cw*0702 were associated with an increased risk for AD (OR 2.3; 95% CI; 1.4-3.7; p=0.001), but not with AD onset age, in a case-control Caucasian population study, annually followed-up for up to 15 years. HLA-B7 and HLA-Cw*0702 being in tight linkage disequilibrium, the true risk loci could not be determined(52).
HLA-B8 was found to be increased in volunteers with impaired cognition. HLA-B8 is part of the so-called “autoimmune” ancestral haplotype (HLA-DQ2, HLA-DR3, HLA-B8, HLA-Cw7 and HLA-A1) which is carried by most Caucasians and known to be positively associated with autoimmune disorders and by elevated circulating levels of inflammatory cytokines, such as IL-1 and TNF‑a(53). HLA B16 was found to be associated with AD in a Japanese study(54). Newer studies found HLA B*07:00/x genotype being a risk factor for sporadic LOAD, in an Iranian study(55). Furthermore, cases with this genotype were found to have worse clinical response at two-year follow-up after rivastigmine treatment than in non-carriers(55).
Regarding non-classical HLA, HFE (homeostatic iron regulator) SNP rs1800562 correlated with slower atrophy rate of the right middle temporal lobe in a study sample obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) that included 812 participants (281 health controls, 483 participants in the mild cognitive impairment stage, and 48 AD participants; Pc=0.003)(56).
HLA class II and AD
In the central nervous system, HLA class II are highly expressed in the microglia, and with lower levels in astrocytes and endothelial cells(39). Both their predisposing and protective role in the AD pathophysiology have been investigated in the recent years.
The first gene of this class that was found to be a risk factor for AD by a large genome-wide association study (GWAS). European ancestry based meta-analysis was the HLA-DRB5-DRB1 region(28). HLA-DRB1 and HLA-DRB5 expressed in the microglia was found to be positively correlated with measures of AD pathology(57).
HLA-DR1, HLA-DR2 and HLA-DR3 were associated with a high risk of AD in two studies: one case-control study of brain tissues of 78 patients with late-onset AD and 50 controls, that found even higher significance in ApoE4 (-)(58), respectively one study based on ADNI data(59).
HLA-DPB1(60) as well as HLA-DRA(25) were associated with a higher risk of AD in two GWAS.
HLA-DRB1*15 was found to be a risk factor for AD (OR 5.4; 95% CI; 2.7-10.8) in a Tunisian population study, as well as DRB1*04 (OR 1.9; 95% CI; 1-3.4), DQB1*05 (OR 0.36; 95% CI; 0.18-0.76), DQB1*06 (OR 3.8; 95% CI; 2.2-6.8), and haplotypes DRB1*1501/DQB1*0602 (OR 5.4; 95% CI; 2.7-10.9) and DRB1*0402/DQB1*0302 (OR 2.9; 95% CI; 1.4-5.7)(61). HLA-DRB1*03 was found to be a risk factor for late-onset AD in a German population study(62).
HLA-DRB1/DQB1 SNPs rs35445101, rs1130399 and rs28746809 were associated with a smaller volume of the left posterior cingulate cortex, as seen on MRI, and rs2854275 was associated with a larger volume. These were found in mild cognitive impairment (MCI) or cognitively normal stage too, suggesting that these SNPs might play a role in disease development from early stages – i.e., MCI or normal cognitive state(59).
The haplotype A*03:01~B*07:02~DRB1*15:01*DQA1*01:02~DQB1*06:02 was also significantly associated with AD in a 11,690 white individuals multicentric study. Class I and class II components separately remained at a significant association(63).
Microglia HLA-DR immunostaining was found to increase with the clinical progression of AD, in the gray matter of entorhinal cortex and in the hippocampus, based on one postmortem study of brain sections of AD and control cases(64).
On the other hand, some HLA class II have been found to offer protection for AD development. HLA-DR4, DR6 and DR9 were associated with a low risk of AD(33,59). HLA-DR4 proved to protect against high levels of glial fibrillary acidic protein, a specific astrocyte marker in AD hippocampus, in a small sample size post-mortem study(65).
HLA DRB1*13:02 proved to protect against total, cortical and subcortical gray matter volume reduction, but the study sample was rather small, and it consisted of only female participants(37). HLA DRB1*13:02 also proved to protect against neural network variability, that is considered to reflect brain dysfunction, even in the presence of ApoE4 risk allele, but, again, the study sample consisted of only females(66). HLA DRB1*13:01 that differs from HLA DRB1*13:02 by a single nucleotide had mixed results(66). Patients carrying HLA-DRB1*04:00/X genotype had a better clinical outcome at the two-year follow-up after rivastigmine treatment than non-carriers(55).
HLA DQB1*06:00/x genotype proved to be protective against LOAD, the cases with this genotype having a milder severity that non-carriers, and had a better clinical response at the two-year follow-up after rivastigmine treatment(55).
Moreover, it was observed that longer the AD progressed, the more decreased were the levels of HLA class II(67).
Some common HLA associations between AD and autoimmune diseases have been found, proving an overlap of pathogenic pathways. HLA-DRB5 SNPs rs2516049 was found both in AD and psoriasis, rs12679874, rs2570088, rs16980051 and rs2298428 were found in Crohn disease, while rs2280231, rs8055533 and rs7258465 were found in type 1 diabetes, all in large-sample data from GWAS(25). Rs2516049 was also associated with higher neurofibrillary tangle accumulation(25).
HLA class III and AD
TNF-a, one of the main proinflammatory cytokines, proved be a risk factor for AD. It was associated with a higher atrophy of both total brain and hippocampal volume than expected for age(56).
On the other hand, in a study based on ADNI data, TNF-a rs2534672 and rs2395488 were associated with a larger volume of the middle temporal lobe, thus offering protection for AD(56).
RAGE (receptor for advanced glycation end products) rs2070600 was associated with progressive atrophy of the right hippocampus-CA1(56).
Discussion and conclusions
Alzheimer’s disease is the result of normal aging plus abnormal pathological injuries(59). Some of these pathological injuries were found to be inflammation-related, being referred to as “inflammaging”(68) or immunosenescence(68). Increasing evidence shows that neuroinflammation is the primary cause of neurodegeneration(69).
Although it is uncertain whether this inflammatory mechanism can cause neurodegeneration or it is present to remove the debris associated with already ongoing neurodegenerative mechanisms, up to date data state that the former is more likely.
The HLA role of this inflammaging is relatively understudied up to date, but data could fill the gap in the state of the art. Although, because of the polymorphisms of HLA genes, different levels of expressions and differences between ethnic and geographical populations exist, it is difficult to find strong associations(29), some HLA gene variants being established as intermediate risk factors for Alzheimer’s disease (39).
A better understanding of neuroinflammation processes could open the path to new therapeutic approaches, such as inflammation resolution agents(14,70) or immunotherapy(71,72).
Bibliografie
- Institute for Health Metrics and Evaluation (IHME). Findings from the Global Burden of Disease Study 2017. Seattle, WA: IHME, 2018.
- National Institute for Health and Care Excellence. Dementia: assessment, management and support for people living with dementia and their carers (NG97). London; 2018 Jun.
- Coman H, Nemeş B. New therapeutic targets in Alzheimer’s disease. Int J Gerontol. 2017;11(1):2-6.
- Liu Z, Li H, Pan A. Discovery and Validation of key biomarkers based on immune infiltrates in Alzheimer’s disease. Front Genet. 2021;12:658323.
- Stahl SM. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 4th Ed. Cambridge: Cambridge University Press; 2013.
- Kim JH. Genetics of Alzheimer’s Disease. Dement Neurocognitive Disord. 2018;17(4):131-36.
- Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312.
- Chouraki V, Seshadri S. Genetics of Alzheimer’s Disease. Adv Genet. 2014;87:245-94.
- Nikolac Perkovic M, Pivac N. Genetic Markers of Alzheimer’s Disease. In: Kim YK (Eds.). Frontiers in Psychiatry. Advances in Experimental Medicine and Biology, vol 1192. Springer, Singapore, 2019.
- Bird T. Alzheimer Disease Overview. Updated: December 20, 2018. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). University of Washington, Seattle. 1997-2019. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=alzheimer
- Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127:3240–3249.
- Guerriero F, Sgarlata C, Francis M, Maurizi N, Faragli A, Perna S, et al. Neuroinflammation, immune system and Alzheimer disease: searching for the missing link. Aging Clin Exp Res. 2017;29:821-831.
- Fiala M, Restrepo L, Pellegrini M. Immunotherapy of Mild Cognitive Impairment by ω-3 supplementation: why are amyloid-β antibodies and ω-3 not working in clinical trials? J Alzheimers Dis. 2018;62:1012-22.
- Zhu M, Wang X, Sun L, Schultzberg M, Hjorth E. Can inflammation be resolved in Alzheimer’s disease? Ther Adv Neurol Disord. 2018;11:1-16.
- Zhang ZG, Li Y, Ng CT, Song YQ. Inflammation in Alzheimer’s Disease and Molecular Genetics: Recent Update. Arch Immunol Ther Exp. 2015;63:333-44.
- Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, Glatzel M. Autoantibodies against beta-amyloid are common in Alzheimer’s disease and help control plaque burden. Ann Neurol. 2009;65:24–31.
- Storace D, Cammarata S, Borghi R, Sanguineti R, Giliberto L, Piccini A et al. Elevation of β-amyloid 1-42 autoantibodies in the blood of amnestic patients with mild cognitive impairment. Arch Neurol. 2010;67:867–872.
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716.
- Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18:759–772.
- Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98:28–41.
- Hampel H, Caraci F, Cuello AC, et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front Immunol. 2020;11:456.
- Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s Disease. Nature. 2020;577:399-404.
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s Disease. J Alzheimer’s Dis. 2010;21:927-938.
- Guerini FR, Tinelli C, Calabrese E, Agliardi C, Zanzottera M, Silvestri A, et al. HLA-A*01 is associated with lade onset of Alzheimer’s disease in Italian patients. Int J Immunoptah Ph. 2009;22(4):991-9.
- Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C,
- et al. Association between genetic traits for immune-mediated disease and Alzheimer’s disease. JAMA Neurol. 2016;73(6):691-7.
- Horton R, Wilming L, Rand V, Lovering R, Bruford EA, Khodiyar VK, et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;5(12):889-99.
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM,
- et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435.
- Lambert JC, Ibrahim-Verbass CA, Harold D, Naj AC, Sims R, Bellenguez C,
- et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452-8.
- Wang ZX, Wan Q, Xing A. HLA in Alzheimer’s Disease: Genetic Association and Possible Pathogenic Roles. Neuromolecular Med. 2020;22:464-473.
- McGeer PL, McGeer EG, Yasojima K. Alzheimer disease and neuroinflammation. J Neural Transm Suppl. 2000;59:53-57.
- Campbell RD, Trowsdale J. Map of the human MHC. Immunol Today. 1993;14:349–52.
- Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature. 1999;401(6756):921-923.
- Misra MK, Damotte V, Hollenbach JA. The immunogenetics of neurological disease. Immunology. 2017;153(4):399-414.
- Mosaad YM. Clinical role oh human leukocyte antigen in health and disease. Scand J Immunol. 2015;82(4):283-306.
- Vică ML, Matei HV, Bondor CI, Nicula GZ, Siserman CV, Loga L, et al. HLA polymorphisms and haplotype diversity in Transylvania, Romania. J Immunol Res. 2019;2019:1342762.
- Robinson J, Halliwell JA, Hayhurst JH, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Research. 2015;43:D423-431.
- James LM, Christova P, Lewis SM, Engdahl BE, Georgopoulos A, Georgopoulos AP. Protective effect of human leukocyte antigen (HLA) allele DRB1*13:02 on age-related brain gray matter volume reduction in healthy women. EBioMedicine. 2018;29:31-7.
- James LM, Georgopoulos AP. Human leukocyte antigen as key factor in preventing dementia and associated apolipoprotein E4 risk. Front Aging Neurosci. 2019;11:82.
- Aliseychik MP, Andreeva TV, Rogaev EI. Immunogenetic factors for neurodegenerative diseases: the role of HLA class II. Biochem (Mosc). 2018;83(9):1385-98.
- Small GW, Ebeling SC, Matsuyama SS, Heyman A, Reisner EG, Renvoize EB,
- et al. Variable association of HLA-A2 in men with early-onset Alzheimer disease. Neurobiol Aging. 1991;12:375-7.
- Small GW, Scott WK, Komo S, Yamaoka LH, Farrer LA, Auerbach SH. No association between the HLA-A2 allele and Alzheimer disease. Neurogenetics. 1999;2:177-182.
- Payami H, Schellenberg GD, Zareparsi S, Kaye J, Sexton GJ, Head MA, et al. Evidence for association of HLA-A2 allele with onset age of Alzheimer’s disease. Neurology. 1997;49:512-8.
- Combarros O, Escribano J, Sanchez-Velasco P, Leyva-Cobian F, Oterino A, Leno C, et al. Association of the HLA-A2 allele with an earlier age of onset of Alzheimer’s disease. Acta Neurol Scand. 1998;98:140-1.
- Harris JM, Cumming AM, Craddock N, St Clair D, Lendon CL. Human leucocyte antigen-A2 increases risk of Alzheimer’s disease but does not affect age of onset in a Scottish population. Neurosci Lett. 2000;294:37-40.
- Zareparsi S, James DM, Kaye JA, Bird TD, Schellenberg GD, Payami H. HLA-A2 homozygosity but not heterozygosity is associated with Alzheimer disease. Neurology. 2002;58:973:5.
- Araria-Goumidi L, Lambert JC, Cottel D, Amouyel P, Chartier-Harlin MC. No association of the HLA-A2 allele with Alzheimer’s disease. Neurosci Lett. 2002;335:75-8.
- Listi F, Candore G, Balistreri CR, Grimaldi MP, Orlando V, Vasto S, et al. Association between the HLA-A1 allele and Alzheimer disease. Rejuv Res. 2006;9(1):99-101.
- Ma SL, Tang NLS, Tam CWC, Lui VWC, Suen EWC, Chiu HFK, et al. Association between HLA-A alleles and Alzheimer’s disease in a southern Chinese community. Dement Geriatr Disord. 2008;26:391-7.
- Guerini FR, Calabrese E, Agliardi C, Zanzottera M, Franceschi M, Grimaldi LME, et al. Association study of the HLA-A2 allele in Italian Alzheimer disease patients. Neurobiol Aging. 2009;30:2082-3.
- Cifuentes RA, Murillo-Rojas J. Alzheimer’s disease and HLA-A2: linking neurodegenerative to immune processes through an in silico approach. Biomed Res Int. 2014:2014:791238
- Wang ZX, Wang HF, Tan L, Sun FR, Tan MS, Tan CC, et al. HLA-A2 alleles mediate Alzheimer’s disease by altering hippocampal volume. Mol Neurobiol. 2017;54:2469:76.
- Lehman DJ, Barnardo MCNM, Fuggle S, Quiroga I, Sutherland A, Warden DR,
- et al. Replication of the association of HLA-B7 with Alzheimer’s disease: a role for homozygosity? J Neuroinflammation. 2006;3:33.
- Esgalhado AJ, Reste-Ferreira D, Albino SE, Sousa A, Amaral AP, Martinho A, et al. CD45RA, CD8β, and IFNγ Are Potential Immune Biomarkers of Human Cognitive Function. Front Immunol. 2020;11:592656.
- Endo H, Yamamoto T, Kuzuya F. HLA system in senile dementia of Alzheimer type and multi-infarct dementia in Japan. Arch Gerontol Geriatr. 1986;5:51-56.
- Rad FR, Akbari MG, Zamani M, Bayat S, Zamani M. Pharmacogenetic and association studies on the influence of HLA alleles and rivastigmine on the Iranian patients with late-onset Alzheimer’s Disease. Mol Neurobiol. 2021;58:2792-2802.
- Wang ZX, Wan Y, Tan L, Liu J, Wang HF, Sun FR, et al. Genetic association of HLA gene variants with MRI brain structure in Alzheimer’s disease. Mol Neurobiol. 2017;54:3195-3204.
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332-337.
- Curran M, Middleton D, Edwardson J, Perry R, McKeith I, Morris C, et al. HLA-DR antigens associated with major genetic risk for late-onset Alzheimer’s disease. NeuroReport. 1997;8:1467–9.
- Wang, ZX, Wang HF, Tan L, Liu J, Wan Y, Sun FR, et al. Effects of HLA-DRB1/DQB1 genetic variants on neuroimaging in healthy, mild cognitive impairment, and Alzheimer’s disease cohorts. Mol Neurobiol. 2017;54:3181-88.
- Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM, et al. Analysis of copy number variation in Alzheimer’s disease: the NIA-LOAD/NCRAD family study. Curr Alzheimer Res. 2012;9(7):801-14.
- Mansouri L, Messalmani M, Klai S, Bedoui I, Derbali H, Gritli N, et al. Association of HLA-DR/DQ polymorphism with Alzheimer’s disease. Am J Med Sci. 2015;349(4):334-7.
- Neill D, Curran MD, Middleton D, Mawhinney H, Edwardson JA, McKeith I, et al. Risk for Alzheimer’s disease in older late-onset cases is associated with HLA-DRB1*03. Neurosci Lett. 1999;275:137-140.
- Steele NZR, Carr JS, Bonham LW, Geier EG, Damotte V, Miller ZA, et al. Fine-mapping of the human leukocyte antigen locus as a risk factor for Alzheimer disease: a case-control study. PLoS Med. 2017;14(3):e1002272.
- Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis Markers. 2006;22:95-102.
- Aisen PS, Luddy A, Durner M, Reinhard Jr JF, Pasinetti GM. HLA-DR4 influences glial activity in Alzheihmer’s disease hippocampus. J Neurol Sci. 1998;161:66-9.
- James LM, Dolan S, Leuthold AC, Engdahl BE, Georgopoulos A, Georgopoulos AP. The effects of human leukocyte antigen DRB1*13 and apolipoprotein E on age-related variability of synchronous neural interactions in healthy women. EBioMedicine. 2018;35:288-94.
- Hoozemans JJ, Rozemuller AJ, van Haastert ES, Eikelenboom P, van Gool WA. Neuroinflammation in Alzheimer’s disease wanes with age.
- J Neourinflammation. 2011;8:171.
- Busse S, Steiner J, Alter J, Dobrowolny H, Mawrin C, Bogerts B, et al. Expression of HLA-DR, CD80, and CD86 in healthy aging and Alzheimer’s disease.
- J Alzheimers Dis. 2015;47:177-84.
- Griciuc A, Tanzi RE. The role of innate immune genes in Alzheimer’s disease. Curr Opin Neurol. 2021;34(2):228–236.
- Medeiros R, Kitazawa M, Passos GF, Baglietto-Vergas D, Cheng D, Cribbs DH, et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease0like pathology in mice. Am J Pathol. 2013;182(5):1780-9.
- Zota V, Nemirovsky A, Baron R, Fisher Y, Selkoe DJ, Altmann DM, et al. HLA-DR alleles in amyloid β-peptide autoimmunity: a highly immunogenic role for the DRB1*1501 allele. J Immunol. 2009;183(5):3522:30.
- Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, et al. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from β pan HLA DR-binding peptide.
- J Immunol. 2005;174:1580-6.

Roadmap to psychiatry. Ghid de psihiatrie
Gabriela Marian
Cartea a fost concepută ca un ghid de psihiatrie destinat tinerei generaţii de studenţi şi rezidenţi în specialitatea Psihiatrie. Apărută la Editura Universitară „Carol Davila” Bucureşti în 2022, a fost tipărită în limba engleză. Cartea se doreşte a fi un instrument prietenos de învăţare a diferitelor aspecte...
Viaţa de student la Medicină – de la stres la depresie şi consum de alcool
Andreea Sălcudean, Andreea-Georgiana Nan, M. Cosma, Bianca-Eugenia Ősz, Virgil Enătescu, Elena‑Gabriela Strete
Primele manifestări ale tulburărilor psihice diagnosticate la adulţi debutează în adolescenţă sau imediat după aceasta, fiind precipitate sau exacerbate de numeroşi factori de stres, precum condiţiile...
Integrarea datelor farmacogenetice în practica psihiatrică
Andrei G. Mangalagiu, B. Petrescu, Cristian A. Cândea, Octavian Vasiliu
Medicina personalizată este un deziderat care prezintă avantajele unei eficacităţi sporite şi ale unei tolerabilităţi superioare, aspecte care ar îmbunătăţi aderenţa terapeutică, calitatea vieţii şi recuperarea funcţiona...
Canabinoidele şi provocările generate de derivaţii lor semisintetici
Ovidiu Alexinschi, Andrei Siriţeanu, Alexandra Boloș
Dintre toate substanţele psihoactive ilicite, canabisul este cel mai consumat drog (4% din populaţie la nivel mondial, anual). În ultimii ani a apărut un nou capitol dedicat canabinoidelor semisintetice, ca răspuns la re...
Ion Vianu
Carla Costescu
Avântul dat de învăţăturile tatălui şi profesorului său, precum şi pasiunea timpurie pentru lectură, „când descoperă bibliotecile publice”, îl iniţiază de la o vârstă fragedă într-o nişă a intelectualilor pasionaţi. ...